
Россия, Троицк


СДЕЛАЙТЕ СВОИ УРОКИ ЕЩЁ ЭФФЕКТИВНЕЕ, А ЖИЗНЬ СВОБОДНЕЕ
Благодаря готовым учебным материалам для работы в классе и дистанционно
Скидки до 50 % на комплекты
только до
Готовые ключевые этапы урока всегда будут у вас под рукой
Организационный момент
Проверка знаний
Объяснение материала
Закрепление изученного
Итоги урока
Была в сети 15.04.2024 16:23

Печерица Наталья Петровна
учитель начальных классов
51 год
Местоположение
Специализация
Болезнь Лея (Ли). Печерица Н.П.
Категория:
Коррекционная школа
19.09.2016 21:10

Просмотр содержимого документа
«Болезнь Лея (Ли). Печерица Н.П.»

Болезнь Лея (Ли)
Выполнила:
Печерица Наталья Петровна, учитель начальных классов
Спец. (корр.) школы-интерната для ОВ с ОВЗ III-IV видов г. Троицка.
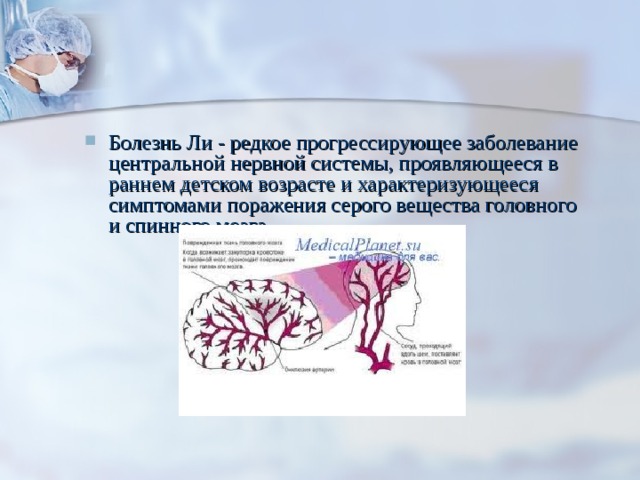
- Болезнь Ли - редкое прогрессирующее заболевание центральной нервной системы, проявляющееся в раннем детском возрасте и характеризующееся симптомами поражения серого вещества головного и спинного мозга.

- О заболевании в первый раз было упомянуто в 1951 г. Тогда
- английского паиологоанатома заинтересовали дети с мышечной
- слабостью, гипотонией и повышенной восприимчивостью к
- инфекциям. Проведя ряд исследований, он выяснил, что
болезнь связана с точечными мутациями митохондриальной ДНК .
- Впервые этот синдром был описан в 1984 году Павлакисом и
- коллегами; десять лет спустя Павлакис и Мицио Хирано
- опубликовали обзор около 100 случаев заболевания.
- К настоящему времени известно более 120 таких случаев.

Болезнь сопровождается полиморфной симптоматикой — диабетом, судорогами, снижением слуха, сердечными заболеваниями, низким ростом, эндокринопатиями, непереносимостью физических нагрузок и нейропсихиатрическими отклонениями. Мутации могут возникать впервые у конкретного пациента, либо наследоваться по материнской линии. Всего к 2009 году было обнаружено 23 миссенсных точечных мутаций и 4 делеции мтДНК, приводящих к синдрому, однако продолжаются сообщения о новых пациентах с симптомами расстройства при отсутствии известных мутаций.

- При данном синдроме происходит аномальное скопление митохондрий - «Рваные (шероховатые) красные мышечные волокна»:

- Это заболевание, наследуемое по материнской линии, чаще возникает в детстве (в том числе раннем), изредка в молодости. Для этой болезни характерны регресс психомоторного развития , эпилептические припадки , дистония, атаксия, атрофия зрительных нервов, офтальмоплегия, тремор, пирамидные симптомы и расстройства дыхания. При КТ и МРТ обнаруживают двусторонние симметричные участки пониженной плотности в базальных ядрах и стволе мозга. Характерен лактацидоз. Течение неуклонно прогрессирующее. Характерно злокачественное течение с летальным исходом. В нескольких семьях в митохондриальном геноме обнаружены точечные мутации в положении 8993 гена 6-й субъединицы H+-АТФ-синтетазы.

- К типичным начальным проявлениям относятся задержка темпа психомоторного развития, дыхательные нарушения. Дети становятся вялыми, сонливыми, снижается мышечный тонус, расстраивается координация движений.

- По мере течения заболевания гипотония мышц может сменяться их спастичиостью, утрачиваются двигательные навыки, появляются миоклонические подергивания мышц. Сухожильные рефлексы снижены или повышены. Могут наблюдаться судороги, глухота, парезы конечностей вследствие поражения периферических нервов. Наряду с этим отмечаются расстройства функции печени, сопровождающиеся снижением аппетита, рвотой, потерей массы тела. У некоторых больных развивается кардиомиопатия.

- Характерны различные зрительные и глазодвигательные расстройства: нарушение цветовосприятия, снижение зрения вплоть до слепоты, атрофия зрительного нерва, нистагм, офтальмоплегия, дискоординированные движения глазных яблок, косоглазие, нарушение реакции зрачков на свет, миоз, мидриаз

- В основе болезни лежит генетически детерминированное нарушение обмена тиамина, наследуемое чаще по аутосомно-рецессивному типу. Вследствие нарушения превращения тиаминпирофосфата в тиаминтрифосфат снижается содержание тиамина во многих отделах ц.н.с. — в стволе мозга, базальных ганглиях, мозжечке, спинном мозге. В печени определяется дефицит пируваткарбоксилазы, что приводит к накоплению пирувата и лактата и нарушениям в цикле трикарбоновых кислот. Морфологические изменения в нервной системе характеризуются поражением серого вещества ствола мозга, базальных ганглиев, четверохолмия, таламуса, мозжечка, спинного мозга.

- Диагноз в амбулаторных условиях затруднен. Предположительный диагноз ставят на основании прогрессирующего нарастания неврологических расстройств, указывающих на избирательное поражение серого вещества головного и спинного мозга и наличия повторных случаев заболевания в семье. Для уточнения диагноза больного необходимо госпитализировать. Примерно у 30% больных в цереброспинальной жидкости обнаруживают увеличение содержания белка и невысокий плеоцитоз лимфоцитарного характера. На ЭКГ выявляют преобладание неспецифической медленноволновой активности.

- Наследуется в соответствии с менделевскими закономерностями, наиболее часто по аутосомно-доминантному типу. Характерный признак этих заболеваний - наличие делеций нескольких участков мтДНК, обусловливающих нарушение структуры и функционирования ряда митохондриальных генов. Механизм возникновения этих нарушений до конца не выяснен. Возможно в его основе лежит возникновение мутаций в ядерных регуляторных генах, контролирующих репликацию мтДНК. Мутации в этих генах могут либо облегчать процесс возникновения перестройки мтДНК, либо снижать активность факторов, распознающих или элиминирующих спонтанно возникающие перестройки.
- В настоящее время картировано три таких гена — в области хромосом 10q 23.3-24, 3р 14.1 -21 и 4q35 — однако, идентифицирован только один - который кодирует фермент аденин-нуклеотид-транслоказу 1, снижение концентрации которой приводит к нарушению метаболизма аденина и нарушению процессов репликации.

- Лечение синдрома пока не известно, зачастую он утягощается, приводя в итоге к гибели пациента. Предпринимаются попытки замедлить его течение. Исследуется применение L-аргинина для уменьшения повреждений мозга при инсультоподобных эпизодах, причём исследователи сообщают, что до принятия препарата уровень аргинина в крови пациентов был значительно снижен во время эпизодов. Предварительные результаты обнадёживают, однако нужны дополнительные исследования. L-аргинин провоцирует выделение NO - газа, имеющего как положительный эффект - расширение сосудов, так и отрицательный: избыток может быть токсичным. Известно, например, что при инфаркте L-аргинин изначально показал положительные результаты, однако более крупные и длительные исследования не подтвердили ранние находки и к тому же выявили повышенную смертность среди тех, кто принимал это средство.
- Также применяется кофермент Q, в отсутствие результатов адекватных клинических исследований; такие исследования начаты, но ещё не завершены.

- Поскольку болезнь может сопровождаться судорогами и не всегда адекватно диагностируется, он может быть принят за «простую» эпилепсию. При этом назначение вальпроата - антиконвульсанта, известного своим негативным воздействием на митохондриальную дыхательную цепь - лишь усугубляет судороги.

- Недавно группа австралийских и американских специалистов заявила, что она нашла генетический дефект, который приводит к развитию синдрома Лея.
- Ученые протестировали более 1000 генов, кодируя протеины, которые демонстрировали активность у двух пациентов. Для анализа генов использовалась новая технология – "секвенирование ДНК нового поколения". Так был выявлен ген, кодирующий энзим, находящийся в митохондриях. Без этого энзима митохондрия не может нормально преобразовывать белки и в итоге развивается синдром. По словам Дэвида Торнберна из Детского исследовательского института Мердока, данное открытие может перевернуть медицину. Новые техники анализа генома позволят в будущем проводить более точную диагностику. Особенно это актуально для пар, уже имеющих больного ребенка и собирающихся заводить второго, ведь на первый взгляд, ребенок рождается здоровым. Однако к трем годам у него появляются проблемы с передвижением, дыханием, грозящие смертью. Просто митохондрии, подпитывающие клетки, отстают по темпам развития.

- Распространённость сложно оценить из-за разнообразия проявлений и связанной с этим трудностью диагностики. Среди взрослого населения Финляндии число лиц с синдромом и мутацией A3243G было оценено в 10.2 человека на 100000. На севере Англии распространённость этой же мутации оценена в 1 на 13000 человек. Как предполагается, митохондриальные заболевания связаны с большой пропорцией нейрогенетических повреждений у взрослых, возможно, являясь одной из наиболее частых причин таких расстройств. Распространённость собственно мутации A3243G, ответственной за большую часть случаев синдрома, оценивается гораздо выше, но мутация не всегда ведёт к заболеванию, поскольку из-за гетероплазмии большинство митохондрий у человека могут быть "здоровыми".

- Рассматриваемым заболеванием страдает, например, Малыш Йозеф из Канады. Врачи Канадского госпиталя уверяли семью в необходимости отключить ребенка от искусственной вентиляции легких,тем самым дать малышу умереть. Врачи отказывались выполнить малышу операцию по наложению трахеостомы, которая могла бы позволить подключить ребенка к аппарату искусственной вентиляции легких через трахеостому на длительный период. Родители не дали умереть малышу и настояли на переводе ребенка в другую больницу.














Вебинар для учителей

Свидетельство об участии БЕСПЛАТНО!
Полезное для учителя

Реализация образовательных программ осуществляется с применением исключительно электронного обучения и ДОТ


