
ПМР, Тирасполь


СДЕЛАЙТЕ СВОИ УРОКИ ЕЩЁ ЭФФЕКТИВНЕЕ, А ЖИЗНЬ СВОБОДНЕЕ
Благодаря готовым учебным материалам для работы в классе и дистанционно
Скидки до 50 % на комплекты
только до
Готовые ключевые этапы урока всегда будут у вас под рукой
Организационный момент
Проверка знаний
Объяснение материала
Закрепление изученного
Итоги урока
Была в сети 18.09.2022 13:14

Мосежная Ангелина Владимировна
учитель биологии и химии
24 года
Местоположение
Специализация
Биология 7 класс
Категория:
Биология
11.05.2022 22:56
Учебник:
Биология. 9 класс. Данилов С.Б., Романова Н.И., Владимирская А.И. - М.: Дрофа, 2017. - 334 с.

Просмотр содержимого документа
«Биология 7 класс»

Наследственные заболевания и их профилактика
Студентка II курса 202 группы
Специальность «Биология»
Мосежная А.
Проверил: Шептицкий В.А.

Этиология наследственных болезней
- Этиологией, то есть причиной наследственных болезней являются мутации. Мутации бывают трех видов:
- генные
- хромосомные
- геномные
- Генные мутации ( точковые мутации)
- Причиной генных мутаций является изменение последовательности нуклеотидов в ДНК, например, добавки, нехватки или перестановки нуклеотидов. Чаще мутирует рецессивный ген, т.к. он неустойчив к неблагоприятным условиям. Такие мутации не проявляются в первом поколении, а накапливаются в генофонде, образуя резерв наследственной изменчивости.
- Генные мутации подвергаются репарации, т.е. удалению мутации гена и восстановлению поврежденной ДНК. Такие мутации самые частые и изменяют фенотип незначительно

- Хромосомные мутации
- Причиной хромосомных мутаций является нарушение структуры хромосомы под действием мутагенных факторов. Различают:
- 1)нехватки: а) концов хромосом ( дефишенси )
- б)середины хромосом ( делеции )
- 2)удвоение (добавки) участков хромосом - дупликации
- 3)повороты участка хромосомы на 180о – инверсии
- 4)перемещение участка внутри хромосомы – транспозиции
- 5)обмен участками между двумя разными хромосомами – транслокации
- Такие мутации сильно изменяют фенотип, т.к. изменяется много генов, не накапливаются в генофонде, т.к. у них очень высокая летальность. Хромосомные мутации могут так же быть материалом для естественного отбора и селекции.
- Делеция по 5 аутосоме (синдром «кошачьего крика») причина - делеция короткого плеча 5 аутосомы. Признаки: у новорожденных нарушение строения гортани, «мяукающий» тембр голоса, слабоумие, отсталость психомоторик
- Делеция хромосомы 21 – хроническое белокровие (лейкемия)
- Синдром «дупликация-делеция 3 аутосомы» - спонтанные аборты, в случае рождения дети не способны сидеть, есть твердую пищу, имеют очень короткий нос.

- Геномные мутации
- Причиной геномных мутаций является изменение числа хромосом в клетке. Они вызывают очень сильные изменения в фенотипе, всегда проявляются в первом поколении.
- Различают три вида геномных мутаций:
- 1) Полиплоидия
- 2)Гетероплоидия
- 3)Гаплоидия
- 4)Полиплоидия
- Самые частые мутации – это генные. Один ген мутирует раз в 40 тысяч лет, но генов миллионы, поэтому 5-10% генов - мутантны.
Наследственные болезни и их классификация
- Проблема здоровья людей и генетика тесно взаимосвязаны. Ученые-генетики пытаются ответить на вопрос, почему одни люди подвержены различным заболеваниям, в то время как другие в этих же, или даже худших условиях остаются здоровы. В основном это связано с наследственностью каждого человека, т.е. свойствами его генов, заключенных в хромосомах.
- В последние годы отмечаются быстрые темпы развития генетики человека и медицинской генетики. Это объясняется многими причинами и прежде всего резким увеличением доли наследственной патологии в структуре заболевания и смертности населения. Статистика показывает, что из 1000 новорожденных у 35-40 выявляются различные типы наследственных болезней, а в смертности детей в возрасте до 5 лет хромосомные болезни составляют 2-3%, генные - 8-10%, мультифакториальные - 35-40%. Ежегодно в нашей стране рождается 180 тысяч детей с наследственными заболеваниями. Более половины из них имеют врожденные пороки, около 35 тысяч - хромосомные болезни и свыше 35 тысяч - генные болезни. Следует отметить, что число наследственных болезней у человека с каждым годом растет, отмечаются новые формы наследственной патологии. В 1956 году было известно 700 форм наследственных заболеваний, а к 1986 году их число увеличилось до 2000. В 1992 году количество наследственных болезней и признаков возросло до 5710.

Хромосомные болезни
- Хромосомные болезни, или синдромы - это группа врожденных патологических состояний, проявляющихся множественными пороками развития, различающихся по своей клинической картине, часто сопровождающихся тяжелыми нарушениями психического и соматического развития. Основной дефект - различные степени интеллектуальной недостаточности, что может осложняться нарушениями зрения, слуха, опорно-двигательного аппарата, более выраженными, чем интеллектуальный дефект, расстройствами речи, эмоциональной сферы и поведения.
- Диагностические признаки хромосомных синдромов можно разделить на три группы:
- неспецифические, т.е. такие, как выраженная умственная отсталость, сочетающаяся с дисплазиями, врожденными пороками развития и черепно-лицевыми аномалиями;
- признаки, характерные для отдельных синдромов;
- патогномоничные для конкретного синдрома, например, специфический плач при синдроме «кошачьего крика».

Хромосомные заболевания не подчиняются менделеевским закономерностям передачи заболевания потомству и в большинстве случаев обнаруживаются спорадически, являясь следствием мутации в половой клетке одного из родителей.
Хромосомные болезни могут быть унаследованы, если мутация имеется во всех клетках родительского организма.
К механизмам, лежащим в основе геномных мутаций, относятся:
- нерасхождение - хромосомы, которые должны были разделяться во время клеточного деления, остаются соединенными и относятся к одному полюсу;
- «анафазное отставание» - утрата отдельной хромосомы (моносомия) может иметь место во время анафазы, когда одна хромосома может отстать от остальных;
- полиплоидизация - в каждой клетке геном представлен более чем дважды.

- Факторы, повышающие риск рождения детей с хромосомными болезнями
- Причины возникновения хромосомных болезней до настоящего времени недостаточно изучены. Имеются экспериментальные данные о влиянии на мутационный процесс таких факторов, как: действие ионизирующих излучении, химических веществ, вирусов. Другими причинами нерасхождения хромосом могут быть: сезонность, возраст отца и матери, порядок рождения детей, прием лекарств во время беременности, гормональные нарушения, алкоголизм и др. Не исключается до определенной степени и генетическое детерминирование нерасхождения хромосом. Повторим, однако, что причины образования геномных и хромосомных мутаций на ранних стадиях развития зародыша до сих пор окончательно не раскрыты.

- К биологическим факторам повышения риска рождения детей с хромосомными аномалиями может быть отнесен возраст матери. Риск рождения больного ребенка особенно резко возрастает после 35 лет. Это характерно для любых хромосомных болезней, но наиболее четко наблюдается для болезни Дауна.
- В медико-генетическом планировании беременности особое значение уделяется двум факторам — наличию анеуплоидии по аутосомам у ребенка и возрасту матери старше 35 лет.
- К кариотипическим факторам риска у супружеских пар относятся: анеуплоидия (чаще в мозаичной форме), робертсоновские транслокации (слияние двух телоцентрических хромосом в области деления) кольцевые хромосомы, инверсии. Степень повышения риска зависит от типа хромосомных нарушений.

Современная проблематика и перспективы
- Современное состояние науки о наследственности не дает никаких оснований для безучастного наблюдения над проявлением тяжелых наследственных пороков у человека, как это имело место еще недавно. Однако сегодня ученым удалось выяснить только связь между нарушениями хромосомного аппарата, с одной стороны, с различными патологическими изменениями в организме человека – с другой. Касаясь вопроса о завтрашнем дне медицинской генетики, можно сказать, что установление взаимосвязи между наследственными заболеваниями и хромосомными повреждениями представляет для клинической медицины большой практический интерес. Выявление причин первоначальных нарушений в системе хромосом, а так же изучение механизма развития хромосомных болезней – также задача ближайшего будущего, причем задача первостепенного значения, так как именно от ее решения во многом зависит разработка эффективных способов профилактики и лечения хромосомных заболеваний.

- Не менее важно для медицинской генетики изучение биохимических сдвигов в организме, возникающих в связи с теми или иными изменениями хромосомного набора. Это поможет установить, какая именно хромосома, или даже какой ее определенный участок контролирует тот или иной процесс обмена веществ. Перед цитогенетикой стоит еще более увлекательная обширная задача, ибо она стремится составить генетическую карту человека, то есть установить расположение генов в хромосомах.
- Дальнейшее изучение взаимосвязи между наследственными заболеваниями и нарушениями в системе хромосом представляет собой чрезвычайно плодотворный синтез теории и практики, в котором генетика обогащается фактическим материалом, а клиническая медицина – знанием тончайших механизмов возникновения различных болезней человека – от тяжелых внутриутробных уродств до злокачественных опухолей, преждевременно прерывающих жизнь взрослого организма.
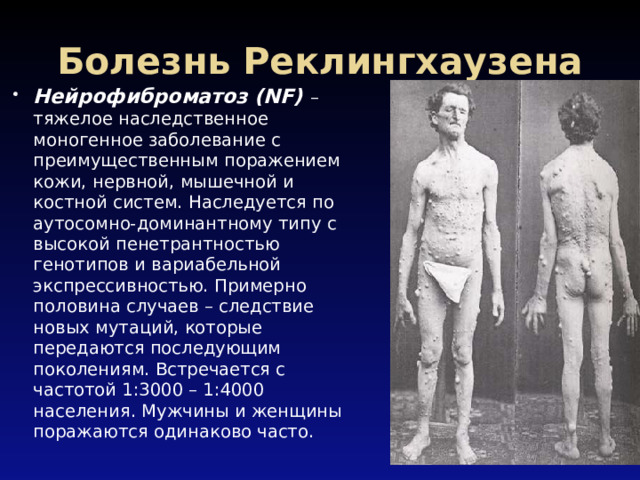
Болезнь Реклингхаузена
- Нейрофиброматоз (NF) – тяжелое наследственное моногенное заболевание с преимущественным поражением кожи, нервной, мышечной и костной систем. Наследуется по аутосомно-доминантному типу с высокой пенетрантностью генотипов и вариабельной экспрессивностью. Примерно половина случаев – следствие новых мутаций, которые передаются последующим поколениям. Встречается с частотой 1:3000 – 1:4000 населения. Мужчины и женщины поражаются одинаково часто.

Нейрофиброматоз
НФ тип 1 – Болезнь Реклингхаузена; периферический НФ
НФ тип 2 – центральный НФ
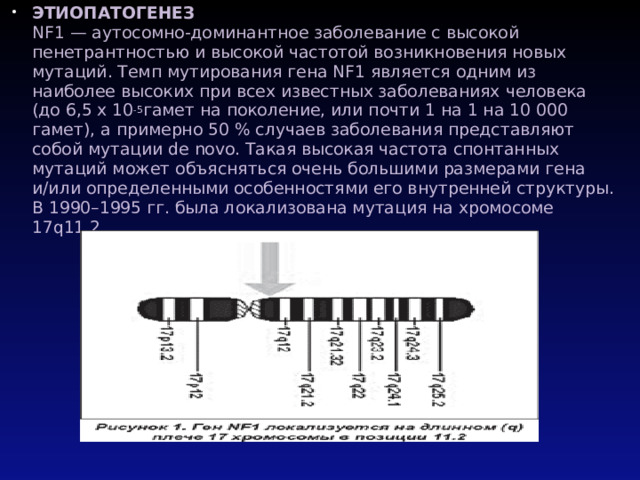
- ЭТИОПАТОГЕНЕЗ NF1 — аутосомно-доминантное заболевание с высокой пенетрантностью и высокой частотой возникновения новых мутаций. Темп мутирования гена NF1 является одним из наиболее высоких при всех известных заболеваниях человека (до 6,5 х 10 -5 гамет на поколение, или почти 1 на 1 на 10 000 гамет), а примерно 50 % случаев заболевания представляют собой мутации de novo. Такая высокая частота спонтанных мутаций может объясняться очень большими размерами гена и/или определенными особенностями его внутренней структуры. В 1990–1995 гг. была локализована мутация на хромосоме 17q11.2.

Особенности течения болезни
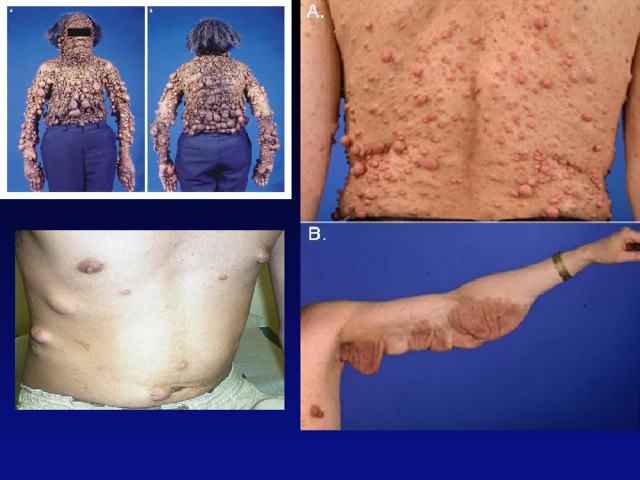
- Заболевание проявляется множественными нейрофибромами по ходу периферических нервов, которые определяются в виде болезненных округлых узелков в толще кожи, варьирующих по своим размерам и локализации. Выявляемость кожного нейрофибоматоза зависит от возраста больных: до 10 лет — 14 %, от 10 до 19 лет — 44 %, 20–29 лет — 85 %, старше 30 лет — 94 %. Чаще первые видимые нейрофибромы появляются в период препубертата или пубертата. К 30-летнему возрасту отмечается неуклонный медленный рост нейрофибром, особенно заметный в период полового созревания индивидуума, а также в период беременности у женщин. После чего рост нейрофибром относительно стабилизируется. Опухоли имеют округлую форму, вариабельные размеры (от просяного зерна до 5 см и более). При пальпации они часто безболезненны, но если в патологический процесс вовлечены периферические нервы, то возникают боли, гипестезии. Опухоль не смещается продольно, а только в поперечном направлении вместе с нервным стволом.

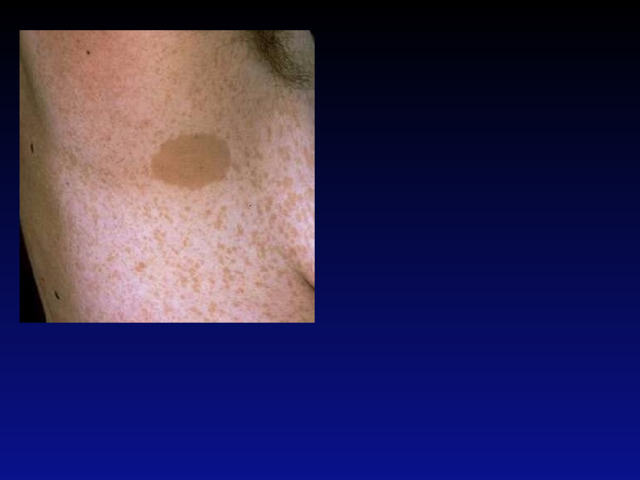

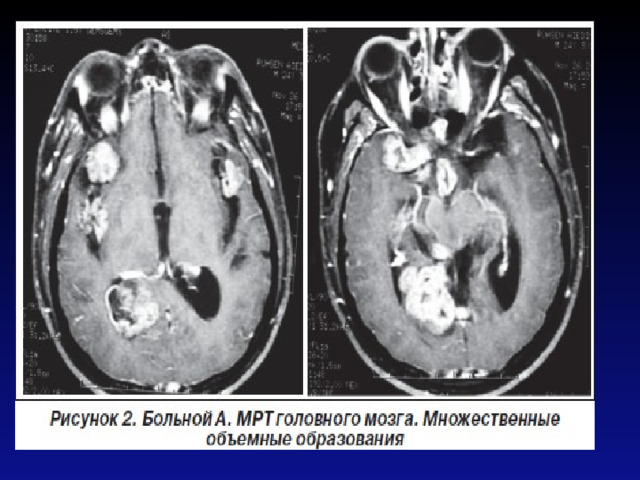
- Первичная диагностика NF должна осуществляться педиатрами, подростковыми врачами, участковыми врачами и врачами общей практики (семейными врачами), а также узкими специалистами (неврологами, дерматологами, офтальмологами, хирургами, стоматологами) в процессе динамического диспансерного обслуживания населения. Важно помнить, что процесс развития клинической симптоматики NF является динамическим, поэтому важны преемственность между специалистами различного профиля и своевременное проведение комплекса дополнительных методов диагностики, включая КТ/МРТ головного и спинного мозга.

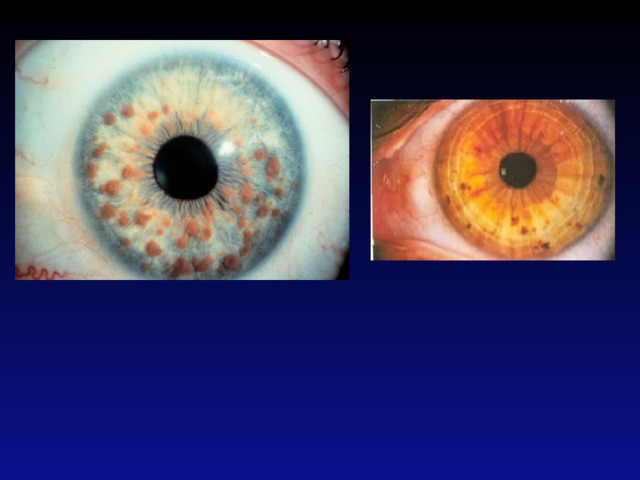
- При постановке диагноза NF1 рекомендуется использовать диагностические критерии. Согласно этим критериям, NF1 может быть диагностирован при наличии у больного не менее 2 из следующих признаков:
- Не менее 5 пигментных пятен цвета «кофе с молоком» диаметром более 5 мм у детей допубертатного возраста и не менее 6 пятен диаметром более 15 мм в постпубертатном возрасте;
- Две и более нейрофибромы любого типа или одна плексиформная нейрофиброма;
- Веснушчатость в подмышечных или паховых складках;
- Дисплазия крыла клиновидной кости или врожденное истончение кортикального слоя длинных костей с псевдоартрозом или без него;
- Глиома зрительного нерва;
- Два и более узелка Лиша на радужке при исследовании с помощью щелевой лампы;
- Наличие у родственников первой степени родства NF1 по тем же критериям.

- В настоящее время не разработано специфической (патогенетической) терапии NF . При нарушениях обучаемости и когнитивных нарушениях рекомендуется обучение детей и подростков в спецшколах и проведение социальной реабилитации пациентов. Опухоли являются причиной болевого синдрома и снижения функций. При выраженном болевом синдроме назначаются НПВС, неопиодные и опиоидные анальгетики, трициклические антидепрессанты (осторожно, ввиду риска провокации судорожного синдрома), топирамат, нейронтин (габапентин). Ортопедические операции показаны при наличии костных деформаций, сколиоза. Хирургические операции также проводятся при наличии болезненных нейрофибром, липом или папиллом больших размеров, а также при расположении опухолей в областях постоянной травматизации или опухолей, являющихся причиной косметического дефекта. Лучевая терапия и химиотерапия проводится в случаях малигнизации опухолей (от 3 до 20 % всех случаев NF).

- Благоприятный прогноз для больных связан с возможностью ранней диагностики злокачественной трансформации и своевременного лечения.
- Следует помнить, что больные NF имеют высокий риск развития серьезных опухолей (в том числе высокий риск малигнизации), которые значительно сокращают продолжительность жизни этих людей. Когнитивные нарушения обычно умеренные, но патогмоничным расстройством является синдром дефицита внимания с гиперактивностью (СДВГ). При получении образования, соответствующего уровню когнитивных функций и IQ, и при рациональном трудоустройстве больные NF могут вести вполне нормальную жизнь. У некоторых больных из-за появления сотен опухолей на коже и стигматизации развивается депрессивный синдром .
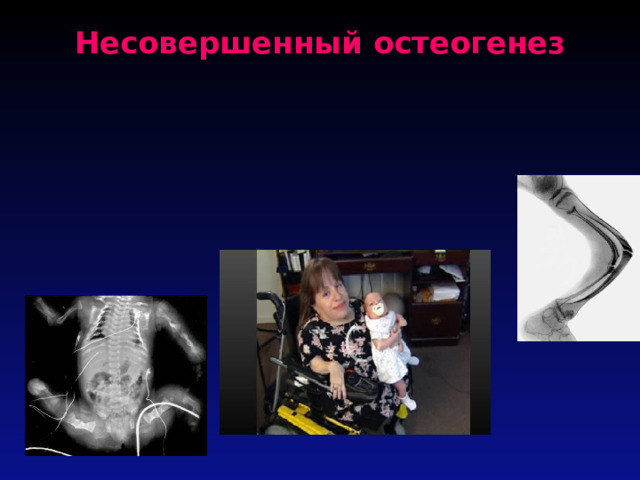
Несовершенный остеогенез

- Несоверше́нный остеогене́з ( лат. osteogenesis imperfecta ) — группа генетических нарушений. Одно из заболеваний ломкости костей. Люди с НО либо имеют недостаточное количество коллагена , либо его качество не соответствует норме. Так как коллаген важный белок в структуре кости, это заболевание влечёт за собой слабые или ломкие кости. Будучи генетическим нарушением, НО является аутосомно -доминантным дефектом. В большинстве переданным по наследству от родителей, однако, возможна и индивидуальная спонтанная мутация .

- Типы Существует четыре типа НО, однако, симптомы варьируют от человека к человеку. 1-й тип наиболее частая и лёгкая форма, за которой следуют 2-й, 3-й и 4-й типы. Сравнительно недавно были классифицированы типы 5-й и 6-й, которые разделяют те же клинические особенности что и 4-й, но каждый из них имеет уникальные гистологические данные.

- 2-й тип Коллаген недостаточного количества или качества. Большинство случаев умирает на протяжении первого года жизни по причине дыхательной недостаточности или внутричерепного кровоизлияния, трудности с дыханием в связи с недоразвитыми лёгкими, тяжёлые деформации кости и невысокий рост. 2-й тип может быть далее разбит на подклассы A, B, C, различаемые радиографическим анализом длинной трубчатой кости и рёбер.

3-й тип Коллаген в достаточных количествах, но недостаточного качества. Кости ломаются легко, иногда даже при рождении, деформация костей, часто тяжёлые, возможны проблемы с дыханием, невысокий рост, искривление позвоночника , иногда также бочковидная грудная клетка, слабость связочного аппарата суставов, слабый мускульный тонус в руках и ногах, обесцвечивание склер (глазных белков), иногда ранняя потеря волос. 3-й тип выделяется из других классификаций будучи типом «Прогрессивной деформации», где новорожденный представляет лёгкие симптомы при рождении и развивает вышеуказанные симптомы в процессе жизни. Продолжительность жизни может быть нормальной, хотя и с тяжёлыми физическими препятствиями .
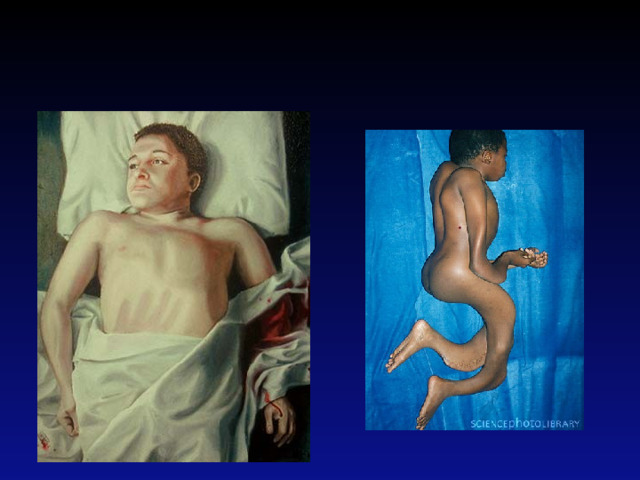

Фенилкетонурия

- это наследственное заболевание обмена одной из важных аминокислот (фенилаланина), в связи с недостатком или полным отсутствием необходимого для обмена фермента
Это приводит к накоплению в организме особо токсичных веществ, поражающих нервную систему
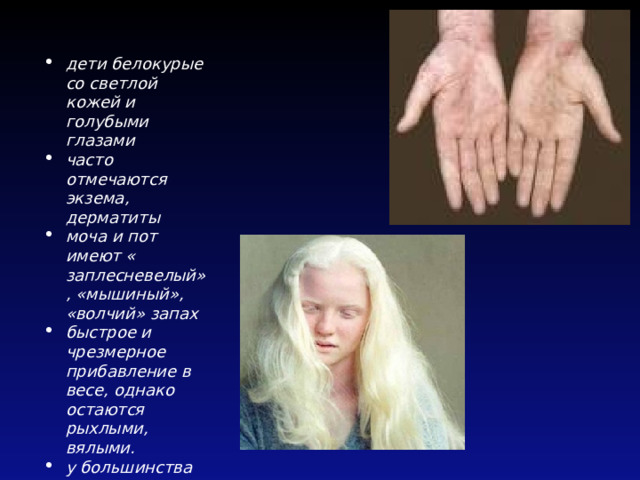
- дети белокурые со светлой кожей и голубыми глазами
- часто отмечаются экзема, дерматиты
- моча и пот имеют « заплесневелый», «мышиный», «волчий» запах
- быстрое и чрезмерное прибавление в весе, однако остаются рыхлыми, вялыми.
- у большинства рано зарастает большой родничок

Неврологическая симптоматика:
- Эпилептиформные припадки
- Нарушение мышечного тонуса
- Плохая координация движений
- Много стереотипии, часты другие знаки экстрапирамидной недостаточности (атетоидные, хореиформные движения)

Расстройства поведения:
- Двигательное беспокойство, целенаправленные, неуправляемые перемещения от объекта к объекту, бесцельные манипуляции с предметами.
или
- Дети пассивны, вялы, плохо узнают близких, оживляются при упоминании о еде.
Профилактика наследственных болезней
- К профилактическим мероприятиям относятся медико-генетические консультации, пренатальная диагностика и диспансеризация. Специалисты во многих случаях могут указать родителям на вероятность появления ребенка с определенными пороками, хромосомной болезнью или нарушениями обмена, обусловленными генными мутациями.
- Медико-генетическое консультирование. Тенденция к увеличению веса наследственной и наследственно обусловленной патологии выражена достаточно четко. Результаты популяционных исследований последних лет показали, что в среднем у 7-8% новорожденных выявляется какая-либо наследственная патология или пороки развития. Самым лучшим методом излечения наследственной болезни было бы исправление патологической мутации путем нормализации хромосомной или генной структуры. Эксперименты по «обратной мутации» проводятся только в микроорганизмах. Однако возможно, что в будущем генная инженерия будет исправлять ошибки природы и у человека. Пока основным способом борьбы с наследственными болезнями являются изменения условий окружающей среды, в результате чего развитие патологической наследственности становится менее вероятным, и профилактика путем медико-генетического консультирования населения.

- Основная цель медико-генетического консультирования – снижение частоты заболеваний путем ограничения появления потомства с наследственной патологией. А для этого необходимо не только установить степень риска рождения больного ребенка в семьях с отягощенной наследственностью, но и помочь будущим родителям правильно оценить степень реальной опасности.
- Направлению в медико-генетическую консультацию подлежат:
- 1) больные с наследственными заболеваниями и члены их семей;
- 2) члены семей, в которых имеются повторные случаи заболевания невыясненной причины;
- 3) дети с пороками развития при подозрении на хромосомные нарушения;
- 4) родители детей с установленными хромосомными нарушениями;
- 5) супруги при повторных спонтанных абортах и бесплодных браках;
- 6) больные с нарушением полового развития
- 7) лица, желающие вступить в брак, если один из них или кто-то из их родственников страдает наследственными заболеваниями.
- В медико-генетической консультации проводиться осмотр больного и составляется родословная семьи. На основании полученных данных предполагается тип наследования данного заболевания. В дальнейшем диагноз уточняется или при исследовании хромосомного набора (в цитогенетической лаборатории), или с помощью специальных биохимических исследований (в биохимической лаборатории).
- При заболеваниях с наследственным предрасположением задача медико-генетического консультирования состоит не в прогнозировании заболевания у потомства, а в определении возможности развития данного заболевания у родственников больного и разработке рекомендации в случае необходимости лечения или соответствующих профилактических мероприятий. Ранняя профилактика, направленная на устранение вредных факторов, провоцирующих развитие заболевания, имеет огромное значение, особенно при высокой степени предрасположенности. К заболевания, при которых такие профилактические мероприятия оказываются действенными, в первую очередь относится гипертоническая болезнь с ее осложнениями, ишемическая болезнь сердца и инсульты, язвенная болезнь, сахарный диабет.

Спасибо за внимание!!!








© 2022, Мосежная Ангелина Владимировна 316 0
Рекомендуем курсы ПК и ППК для учителей





Похожие файлы
Вебинар для учителей

Свидетельство об участии БЕСПЛАТНО!
Полезное для учителя

Реализация образовательных программ осуществляется с применением исключительно электронного обучения и ДОТ



