
Россия, Нижний Новгород


СДЕЛАЙТЕ СВОИ УРОКИ ЕЩЁ ЭФФЕКТИВНЕЕ, А ЖИЗНЬ СВОБОДНЕЕ
Благодаря готовым учебным материалам для работы в классе и дистанционно
Скидки до 50 % на комплекты
только до
Готовые ключевые этапы урока всегда будут у вас под рукой
Организационный момент
Проверка знаний
Объяснение материала
Закрепление изученного
Итоги урока
Была в сети 16.09.2023 02:09

Билялова Динара Рамильевна
Учитель начальных классов
25 лет
Местоположение
Специализация
Методическая разработка на тему: "Наследственные заболевания обмена веществ как причина сложных дефектов.".
Категория:
Коррекционная школа
24.07.2021 18:15

Просмотр содержимого документа
«Методическая разработка на тему: "Наследственные заболевания обмена веществ как причина сложных дефектов.".»
МИНОБРНАУКИ РОССИИ
ФЕДЕРАЛЬНОЕ ГОСУДАРСТВЕННОЕ БЮДЖЕТНОЕ ОБРАЗОВАТЕЛЬНОЕ УЧРЕЖДЕНИЕ
ВЫСШЕГО ОБРАЗОВАНИЯ
«НИЖЕГОРОДСКИЙ ГОСУДАРСТВЕННЫЙ ПЕДАГОГИЧЕСКИЙ УНИВЕРСИТЕТ ИМЕНИ КОЗЬМЫ МИНИНА»
Факультет психологии и педагогики
Кафедра специальной педагогики и психологии
Направление подготовки: 44.03.03 Специальное (дефектологическое) образование
Профиль подготовки: Олигофренопедагогика
Реферат
На тему: Наследственные заболевания обмена веществ как причина сложных дефектов.
Билялова Динара Рамильевна
Нижний Новгород – 2020 г
Вступление 3
Глава 1. Теоретический обзор проблемы наследственных заболеваний обмена веществ как причина сложных дефектов. 4
1.1. Наследственные заболевания 4
1.2. Сложные дефекты 6
1.3. Наследственные заболевания обмена веществ как причина сложных дефектов. 8
Глава 2. Практическая часть. 9
2.1. Основные формы НБО. 9
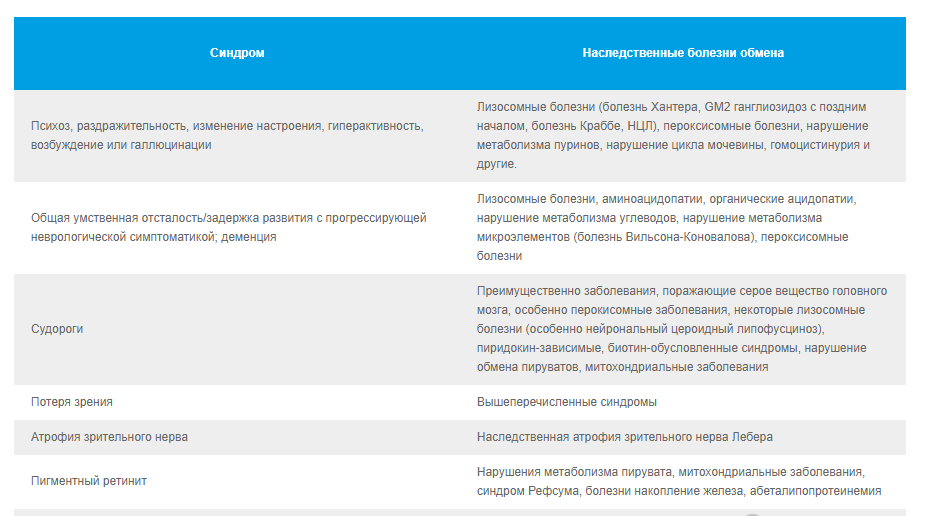
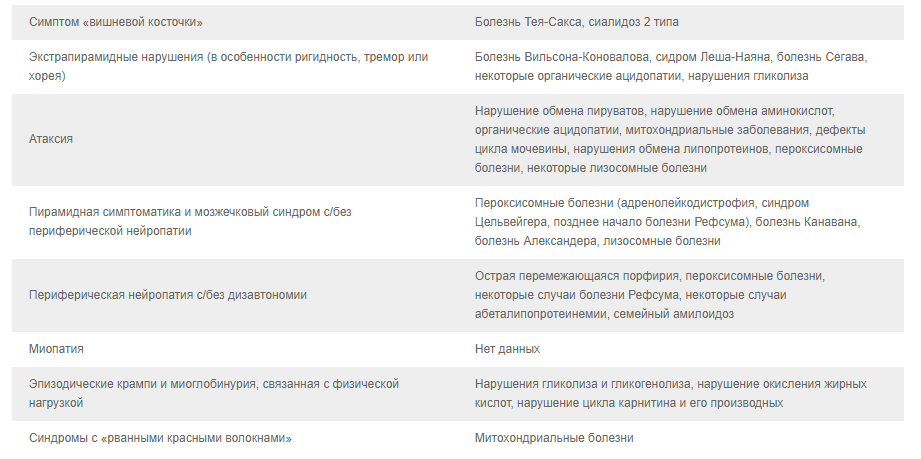
2.2. Классические неврологические синдромы, ассоциированные с известными НБО. 10
2.3. Преимущества генетического исследования заболеваний 11
2.4. Лечение 12
Заключение 13
Список используемой литературы: 15
Приложения 16
Большинство наследственных заболеваний обмена веществ (также называемых врожденными дефектами метаболизма) обусловлены мутациями в генах, кодирующих ферменты; дефицит фермента или его неактивность приводят к накоплению субстрата или метаболитов, или к недостатку производных фермента. Такие заболевание являются актуальными в современном мире, потому что идентифицированы сотни таких расстройств, и, хотя большинство наследственных нарушений обмена веществ крайне редки по отдельности, в целом они представляют довольно распространенную группу расстройств.
Наследственные метаболические нарушения обычно группируются в зависимости от пораженного субстрата, например:
Нарушения обмена аминокислот
Нарушения углеводного обмена
Нарушения метаболизма жирных кислот
Нарушения метаболизма пуринов и пиримидинов
В большинстве стран обязательным является проведение неонатального скрининга всех новорожденных на наличие конкретных наследственных нарушений обмена веществ и других состояний, включая фенилкетонурию, тирозинемию, недостаточность биотинидазы, гомоцистинурию, болезнь кленового сиропа и галактоземию. Во многих государствах имеются расширенные скрининговые программы, которые охватывают множество врожденных нарушений мfетаболизма, включая нарушения окисления жирных кислот и другие органические ацидемии. Исчерпывающий обзор каждого из этих состояний см. в техническом отчёте "Листы и таблицы алгоритмов и аналитика состояния скрининга новорожденных" (newborn screening ACT sheets and algorithm table) Американской коллегии медицинской генетики и геномики (ACMG).
Нарушения обмена веществ, в основном вызывающие заболевания у взрослых (например, подагра, порфирия), органоспецифичные заболевания (например, болезнь Вильсона, врожденная гиперплазия надпочечников) или являются общими (например, кистозный фиброз, гемохроматоз), обсуждаются в других разделах Руководства. Для наследственных заболеваний метаболизма липопротеинов – Генетическая (Первичная) Дислипидемия.
- Наследственные заболевания
Наследственные нарушения обмена веществ включают в себя большую группу наследственных заболеваний, затрагивающих расстройства метаболизма. Такие нарушения составляют значительную часть группы метаболических расстройств (метаболические заболевания).
Метаболические заболевания — гетерогенная группа болезней, при которых нормальные метаболические процессы в тканях нарушены, чаще всего из-за отсутствия или недостаточности определённого фермента, и, как следствие, патологического накопления веществ, обладающих токсическим действием или нарушающих способность синтеза других жизненно важных соединений.
Типичные симптомы полиморфны и зачастую вовлекают несколько систем организма:
- Задержка роста, психо-моторного развития, потеря веса
- Недоразвитые гениталии, задержка или преждевременное половое созревание
- Задержка развития, судороги, деменция, энцефалопатия, инсульты
- Глухота, слепота, нарушение восприятия боли
- Кожная сыпь, гиперпигментация, недержание пигмента, избыточное оволосенение
- Аномалии развития зубов
- Иммунодефицит, тромбоцитопения, анемия, увеличенная селезенка, увеличенные лимфоузлы
- Повторяющаяся рвота, диарея, абдоминальные боли
- Повышение диуреза, нарушения функции почек, дегидратация, отеки
- Гепатомегалия, нарушение функции печени
- Малые аномалии развития (стигмы дизэмбриогенеза), врожденные пороки развития
- Тахипноэ, нарушения дыхания
- Нарушение поведения, депрессия, психозы
- Боль в суставах, мышечная слабость, крампи
В большинстве случаев врожденные нарушения метаболизма проявляются в первые дни жизни. Однако, они могут остаться нераспознанными в период новорожденности, и диагноз может быть поставлен только через несколько месяцев и даже лет, или же в редких случаях дебютировать в более позднем возрасте.
Врожденное нарушение обмена веществ должно рассматриваться, как возможное состояние, у любого ребенка с одним или более из указанных клинических проявлений:
1) необъяснимое отставание умственного, двигательного развития, судороги;
2) повышенный уровень определенных метаболитов в крови или моче (например, при исследованиях мочи на органические ацидурии или крови с помощью тандемной масс-спектрометрии)
3) необычный запах, в частности во время острого заболевания;
4) интермиттирующие эпизоды необъяснимой рвоты, ацидоза, нарушений психики, кома;
5) гепатомегалия;
6) почечные камни.
В данную панель входят такие группы заболеваний как:
- нарушения обменов углеводов (например, болезни накопления гликогена),
- аминоацидопатии (например, фенилкетонурия),
- органические ацидурии (например, алькаптонурия),
- нарушения окисления жирных кислот (например, глютаровая ацидемия 2 типа),
- митохондриальные болезни (например, синдром Кернс-Сейера),
- порфирии (например, острая перемежающаяся порфирия),
- нарушение пуринов или пиримидинов (например, синдром Синдром Лёша-Найхана),
- нарушение обмена стероидов (например, врожденная гиперплазия надпочечников),
- пероксисомные болезни (например, синдром Цельвейгера)
- лизосомные болезни (например, болезнь Гоше),
- и другие заболевания со схожими проявлениями.
- Сложные дефекты
Сложный дефект представляет собой не просто сочетание (сумму) двух и более дефектов развития; он является качественно своеобразным и имеет особую структуру, отличную от составляющих его аномалий.
Категорию детей со сложными дефектами составляют (42):
Дети с умственной отсталостью, отягощенной нарушениями слуха;
Дети с умственной отсталостью, осложненной нарушениями зрения;
Дети глухие слабовидящие;
Слепоглухонемые дети;
Дети с задержкой психического развития, которая сочетается с дефектами зрения или слуха;
Глухие дети с нарушениями соматического характера (врожденные пороки сердца, заболевания почек, печени, желудочно-кишечного тракта).
Кроме того, в дефектологической практике встречаются дети с множественными дефектами. К ним относятся:
1. Дети с умственной осталостью слепоглухие;
2. Дети с нарушениями опорно-двигательного аппарата в сочетании с дефектами органов слуха, зрения, речи или интеллектуальной недостаточностью.
Таким образом, к детям со сложными дефектами можно отнести детей, у которых отмечаются нарушения развития сенсорных и моторных функций в сочетании с недостатками интеллекта (задержка психического развития, умственная отсталость).
Причинами возникновения сложных дефектов могут быть, как указывает Б.П.Пузанов, наследственные и экзогенные факторы.
Наиболее тяжелой группой детей со сложными дефектами выступают слепоглухонемые дети. Её составляют дети, не только полностью лишенные зрения, слуха и речи, но и с парциальным (частичным) поражением сенсорной сферы: слепые с такой потерей слуха, которая препятствует усвоению речи на слух, и глухие с такой потерей зрения, которая препятствует зрительной ориентировке.
Специфической особенностью таких детей является практически полная невозможность получать информацию об окружающем по естественным каналам, что увеличивает значимость коррекционного образования для них, по сравнению с другими детьми, имеющими сложные дефекты. При этом у слепоглухонемого ребенка часто могут быть развиты все усложняющиеся формы общения - от элементарных жестов (воспринимаемых посредством осязания) до вербальной речи. Это позволяет таким детям относительно успешно овладевать программой средней общеобразовательной школы, а некоторым оканчивать высшие учебные заведения.
Патологии, также известные как врожденные нарушения метаболизма, — это заболевания, причина которых кроется в генетическом изменении белка или фермента, в результате чего блокируется определенный процесс метаболизма. Такая блокировка влияет на нормальное функционирование некоторых клеток и органов и проявляется рядом симптомов, различных у каждого пациента. Среди таких симптомов могут встречаться разные виды неврологических синдромов.
Эта группа патологий очень обширна, однако ее можно систематизировать с помощью действующей классификации, которая в данный момент претерпевает значительные изменения ввиду того, что сегодня мы располагаем гораздо большими знаниями о базовых механизмах развития таких патологий. Ниже приведены основные группы патологий, составленные на основании типа поражения организма при каждой из них.
Врожденное нарушение метаболизма малых молекул:
Влияют на промежуточный метаболизм. Сюда входят аминоацидопатии (фенилкетонурия и пропионовая ацидурия). Также сюда входит нарушения обмена углеводов или нейромедиаторов и нейромодуляторов.
Врожденное нарушение энергетического обмена
Характеризуется недостаточной выработкой и использованием энергии. Сюда входят митохондриальные заболевания, недостаточная выработка пирувата или глюкозы (в мышцах или печени) и т. д.
Врожденное нарушение метаболизма сложных молекул:
Группа заболеваний, которые препятствуют синтезу больших молекул. Они проявляются в виде постоянных симптомов, не связанных с питанием. Сюда входят лизосомные (мукополисахаридоз, олигосахаридоз, сфинголипидоз и т. д.), пероксисомальные (синдром Цельвегера, адренолейкодистрофия, сцепленная с хромосомой Х) заболевания и врожденные нарушения гликозилирования, а также другие врожденные нарушения метаболизма.
Мутации в отдельных генах приводят к нарушению синтеза или разрушения белков, углеводов, жиров или сложных веществ. Большинство из них связаны с дефектами ферментов или транспортных белков, в результате чего происходит блок определенного метаболического пути, определенная биохимическая реакция перестает работать и в клетках накапливаются субстраты этих реакций и их производные.
Основная симптоматика проявляется из-за накопления токсических веществ перед блоком, патологических альтернативных путей метаболизма, уменьшения продукции энергетических субстратов или как следствие дефицита конечных продуктов биохимической реакции после блока метаболического пути.
Почти каждое заболевание из этой группы имеет несколько форм, которые различаются по возрасту начала заболевания, клинической выраженности, и, нередко, по типу наследования.
Нарушения обменов углеводов (например, болезни накопления гликогена)
Аминоацидопатии (например, фенилкетонурия)
Органические ацидопатии (например, алькаптонурия)
Нарушения окисления жирных кислот (например, глютаровая ацидемия 2 типа)
Лизосомные болезни (например, болезнь Гоше)
Порфирии (например, острая перемежающаяся порфирия)
Нарушение пуринов или пиримидинов (например, синдром Синдром Лёша-Найхана)
Нарушение обмена стероидов (например, врожденная гиперплазия надпочечников)
Пероксисомные болезни (например, синдром Цельвейгера)
Митохондриальные болезни (например, синдром Кернс-Сейера)
![]()
Генетические исследования позволяют выявлять мутации в известных генах, ассоциированных с НБО. Со временем уже существующие панели генов дополняются и расширяются с учетом мировых баз данных.
![]()
Точный молекулярно-генетический анализ позволяет не проводить дальнейшие диагностические исследования и более точно прогнозировать течение заболевания.
![]()
Анализ проходит быстро и безболезненно. Всё, что требуется для анализа – это образец крови. Специалисты сравнивают результаты со специальной панелью, которая позволяет выявить возможную причину заболевания.
![]()
Пациент получает точные результаты, на основании которых специалисты могут назначить максимально эффективное лечение.
![]()
При проведении процедуры строго соблюдается конфиденциальность.
Большинство наследственных болезней обмена в настоящее время являются неизлечимыми, и терапевтические возможности сводятся к симптоматической и патогенетической терапии: диетотерапия (ограничение поступления в организм определенных веществ), специальные лечебные продукты питания, препараты, направленные на снижение образования токсичных метаболитов, восполнение некоторых метаболитов, которых не хватает в организме, фермент-заместительная терапия.
При некоторых формах НБО (мукополисахаридоз 1 типа, ряд анемий и наследственных нарушениях иммунной системы) эффективно проведение трансплантации гемопоэтических стволовых клеток.
Поэтому важно установить диагноз как можно раньше, чтобы лечение было более эффективно.
Исходя из всего выше сказанного, можно сделать вывод о том, что наследственные нарушения обмена веществ включают в себя большую группу наследственных заболеваний, затрагивающих расстройства метаболизма. Такие нарушения составляют значительную часть группы метаболических расстройств (метаболические заболевания). Наследственными называются такие заболевания человека, которые вызваны перестройками и нарушениями в генетическом материале организма – хромосомах и генах. Среди наследственных заболеваний человека одно из самых значительных мест занимают наследственные болезни обмена. В настоящее время эта группа включает около 700 различных заболеваний
Развитие большинства из них является следствием дефекта единичных генов, кодирующих индивидуальные ферменты, которые обеспечивают превращение одних веществ (субстраты) в другие (продукты). В большинстве случаев таких расстройств патогенным является накопление веществ, обладающих токсическим действием или нарушающих способность синтеза других жизненно важных соединений. Для наследственных болезней обмена веществ иногда используется синонимичный термин «врождённые ошибки метаболизма» ферментопатии.
Talking glossary of genetic terms: genome (англ.). National Human Genome Research Institute. Дата обращения 1 ноября 2012.Архивировано 4 ноября 2012 года.
↑ International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. (англ.) // Nature : journal. — 2004. — Vol. 431, no. 7011. — P. 931—945. [1]
↑ Перейти обратно:1 2 International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. (англ.) // Nature : journal. — 2001. — Vol. 409, no. 6822. — P. 860—921. [2]
↑ «Мусорная» ДНК помогает включать гены.
↑ «Мусорная» ДНК играет важнейшую роль в поддержании целостности генома.
↑ Tjio J. H., Levan A. The chromosome number of man (неопр.) // Hereditas (англ.)русск.. — 1956. — Т. 42. — С. 1—6. Первая работа с точно установленным числом хромосом у человека.
↑ Human Chromosome Number, здесь рассказана история подсчёта хромосом у человека
↑ Nei M., Xu P., Glazko G. Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. (англ.) // Proceedings of the National Academy of Sciences of the United States of America : journal. — 2001. — Vol. 98, no. 5. — P. 2497—2502.
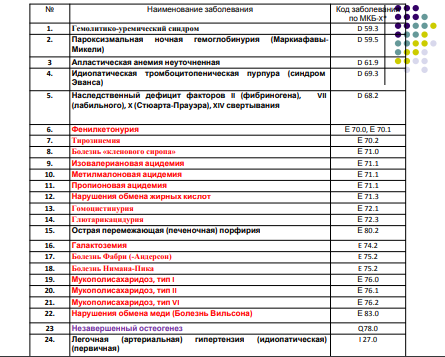
ПРИЛОЖЕНИЕ 1

ПРИЛОЖЕНИЕ 2

ПРИЛОЖЕНИЕ 3














Вебинар для учителей

Свидетельство об участии БЕСПЛАТНО!
Полезное для учителя

Реализация образовательных программ осуществляется с применением исключительно электронного обучения и ДОТ



