
Россия, Петропавловск-Камчатский


СДЕЛАЙТЕ СВОИ УРОКИ ЕЩЁ ЭФФЕКТИВНЕЕ, А ЖИЗНЬ СВОБОДНЕЕ
Благодаря готовым учебным материалам для работы в классе и дистанционно
Скидки до 50 % на комплекты
только до 22.06.2025
Готовые ключевые этапы урока всегда будут у вас под рукой
Организационный момент
Проверка знаний
Объяснение материала
Закрепление изученного
Итоги урока
Была в сети 02.06.2025 01:16
Барышева Любовь Александровна
31 год
Местоположение
Специализация
Методическая разработка лекционного занятия "Наследственные болезни и их классификация"
Категория:
Биология
10.02.2021 13:01

Просмотр содержимого документа
«Методическая разработка лекционного занятия "Наследственные болезни и их классификация"»
Тема: «Наследственные болезни и их классификация. Хромосомные болезни».
Тип учебного занятия: теоретическое занятие
Цели занятия:
1.Образовательные: сформировать знания о видах наследственных болезней человека.
2.Развивающие: формирование у студентов интереса к данной дисциплине умение обобщать материал, делать выводы, а также умений осуществлять поиск и использовать необходимую информацию.
3.Воспитательные: формирование у студента интереса к своей будущей профессии, развитие навыков анализировать, проводить сравнение.
Мотивация
Тема «Хромосомные болезни» является одной из наиболее интересных и основополагающих в процессе изучения медицинской генетики. До XX века главной проблемой медицины были инфекционные заболевания, уносившие миллионы человеческих жизней. Открытие антибиотиков вложило в руки медиков эффективное орудие борьбы со многими инфекциями. В настоящее время медицинским работникам все чаще приходится иметь дело с наследственной патологией, число которой постоянно растет, отмечаются новые формы наследственной патологии. Эти заболевания могут с высокой вероятностью явиться причиной инвалидности или преждевременной гибели пациента. Поэтому в современных условиях лабораторные техники должны иметь представление о различных наследственных патологиях, о причинах их возникновения, возможных мерах предупреждения их возникновения, об особенностях ухода за больными с наследственной патологией. Материал, изученный на данной лекции, может быть использован в будущей работе лабораторного техника для исследования на молекулярном уровне.
Материально-техническое оснащение занятия
1.Презентация: «Наследственные болезни и их классификация. Хромосомные болезни» в электронном виде.
2.Учебная литература: Учебная литература: Акуленко Л.В. Биология с основами медицинской генетики: учеб. для студентов образоват. учреждений сред. проф. образования, обучающихся по специальности 060110.51 «Лаб.диагностика» по дисциплине «Биология с основами мед.генетики» /Л.В. Акуленко, И.В. Угаров; под ред. О.О.Янушевича и С.Д.Арутюнова. - М.: ГЭОТАР-Медиа, 2011. - 368с.
Продолжительность: 80 мин
Место проведения: кабинет № 305
Требования к уровню усвоения учебного материала
После изучения темы студент должен уметь
-
конспектировать излагаемый материал;
-
иметь представление о видах наследственных болезней;
Студент должен знать:
-
Классификацию наследственных болезней.
-
Болезни связанные с нарушением числа хромосом.
-
Болезни связанные с нарушением структуры хромосом.
После занятия у студентов должны сформироваться элементы
следующих компетенций
Ожидаемые результаты обучения
Настоящее теоретическое занятие дает возможность для формирования профессиональных (ПК) и общих компетенций (ОК) студентов
ПК – профессиональные компетенции
Код - Наименование результата обучения
ПК 4.5. Соблюдение правил медицинской этики
ОК – общие компетенции
ОК 1. Понимать сущность и социальную значимость своей будущей профессии, проявлять к ней устойчивый интерес.
ОК 2. Организовывать собственную деятельность, выбирать типовые методы и способы выполнения профессиональных задач, оценивать их выполнение и качество
ОК 3. Принимать решения в стандартных и нестандартных ситуациях и нести за них ответственность
ОК 4. Осуществлять поиск и использование информации, необходимой для эффективного выполнения профессиональных задач, профессионального и личностного развития
ОК 5. Использовать информационно-коммуникационные технологии в профессиональной деятельности.
ОК 6. Работать в коллективе и команде, эффективно общаться с коллегами, руководством, потребителями.
ОК 7. Брать на себя ответственность за работу членов команды (подчиненных), за результат выполнения заданий
ОК 8. Самостоятельно определять задачи профессионального и личностного развития, заниматься самообразованием, осознанно планировать и осуществлять повышение квалификации.
ОК 13. Организовывать рабочее место с соблюдением требований охраны труда, производственной санитарии, инфекционной и противопожарной безопасности.
Междисциплинарные связи
-
Биология
-
Анатомия человека
-
Физиология человека
Внутрипредметные связи:
-
Наследование признаков человека.
-
Изменчивость. Факторы мутагенеза.
-
Внутриутробное развитие человека. Врожденные пороки развития.
-
Методы изучения наследственности и изменчивости человека.
Структурно-логическая схема занятия
«Наследственные болезни и их классификация. Хромосомные болезни»
| Этапы учебного занятия | Время | Действия преподавателя | Действия студента |
| 1.Организационный момент | 5 мин | Проверка присутствующих/отсутствующих, проверка внешнего вида, готовность к занятию | Готовность к занятию: внешний вид, наличие средств обучения |
| 2. Актуализация знаний | 5 мин | Формирование и мотивация темы занятия: 1.Что такое наследственность?
2.Что такое хромосома?
3.Что такое патология?
(Приложение 1) | Студенты формулируют тему занятия |
| 3.Изучение нового материала | 40 мин | Изложение лекционного материала с демонстрацией презентации «Наследственные болезни и их классификация. Хромосомные болезни» | Студенты составляют конспект, фиксируют основные понятия |
| 4.Обобщение и систематизация материала | 15 минут | Преподаватель организовывает беседу по уточнению данной темы: 1.Какова классификация наследственных заболеваний? 2.Дайте определение понятию хромосомная болезнь 3.Перечислите хромосомные заболевания человека, как их можно классифицировать? 4.Что вы знаете о врожденной патологии? 5.Дайте характеристику синдрома Дауна, синдром Эдвардса, синдром Патау, Шерешевского-Тернера, синдром Кошачьего крика, синдром Клайнфельтера.
(Приложение 2) | Студенты обобщают свои знания, делают выводы |
| 5.Подведение итогов | 10 мин | Комментирование результатов усвоения знаний по теме занятия. Ответить на предложенные вопросы для закрепления, изученного материла. Словесное поощрение студентов | Студенты по окончанию занятия задают вопросы по теме лекционного материала |
| 6. Домашнее задание студентам | 5 мин | Преподаватель дает домашнее задание.
1.Составление электронных презентаций по темам (на выбор):
1.«Проявление умственной отсталости при хромосомных синдромах» 2.«Половая функция при хромосомных синдромах» 3.«Группы риска по развитию хромосомных синдромов» 4. «Синдром трисомии 8» 5. «Синдром Вольфа-Хиршхорна» 6.«Синдром кошачьего глаза» 7. «Синдром Реторе» 8.«Синдром Орбели» 9.«Синдром де Груши» 10.Синдром трипло-Х 11.Синдром ХУУ
2.Тест на платформе moodle. (Приложение 3) | Студенты записывают домашнее задание. Изучают пройденный лекционный материал и проходят тестовые задания к лекционному материалу. |
Лекция №9 Наследственные болезни и их классификация. Хромосомные болезни
Генетика и проблемы здоровья тесно связаны между собой. Почему одни люди подвержены различным заболеваниям, а другие находясь даже в худших условиях, здоровы? Выяснено, что связано с наследственностью каждого человека, свойствами его генов, находящихся в хромосомах. По статистике 1 000 новорожденных у 35-40 Выявляются различные типы наследственных болезней. Ежегодно в нашей стране рождается 180 тыс., детей с наследственными заболеваниями. Из них больше половины имеют врожденные пороки, около 35 тыс. - хромосомные болезни и свыше 35 тыс. - генные болезни. В настоящее время медицинским работникам все чаще приходится иметь дело с наследственной патологией, число которой постоянно растет, отмечаются новые формы наследственной патологии. Эти заболевания могут с высокой вероятностью явиться причиной инвалидности или преждевременной гибели пациента.
Наследственные болезни — это патологические состояния, в основе которых изменение наследственного материала (мутация). В развитии таких заболеваний главную роль играют нарушения в структуре гена или хромосомы. В 1956 г. было известно 700 форм наследственных заболеваний, к 1992 г. число их возросло до 5 710. Решающее слово в диагностике наследственных болезней имеют лабораторные и биохимические анализы.
Люди с наследственной патологией нуждаются в постоянной медицинской помощи и социальной защите, поскольку часто являются инвалидами. Практически все разделы клинической медицины включают наследственные болезни. Например, около 70% случаев нарушений зрения 45% тугоухости относятся к этой патологии. Среди нервных болезней выделяют примерно 350 заболеваний, обусловленных генными мутациями, в дерматологии - 250 и т.д. Необходимо отметить, что понятие «врожденные болезни» не является синонимом понятия «наследственные болезни». Врожденная патология выявляется у ребенка при его рождении. Она может быть вызвана не только мутациями, но и одними факторами внешней среды, которые повреждают плод (внутриутробные инфекции, травмы и т.д.).
Наследственные заболевания не всегда про являются с момента рождения или даже в детском возрасте. Некоторые из них (например, хорея Гентингтона) могут проявляться в 40-50 лет. Кроме того, семейные болезни» тоже не всегда являются наследственными, так члены одной семьи обычно попадают под влияние одинаковых факторов внешней среды и могут иметь однотипные патологические нарушения. Термин «синдром» в клинической генетике употребляется только для обозначения совокупности симптомов, имеющих единый патогенез и составляющих самостоятельную нозологическую единицу. Термины «болезнь» и «синдром» для наследственной патологии равнозначны. Любой вид патологии имеет свои закономерности клинического проявления.
Наследственная патология, содержащая огромное нозологическое многообразие, имеет специфические черты, которые необходимо знать в качестве ориентиров в диагностических поисках.
Все наследственные болезни делятся по характеру изменения наследственных структур на три основных типа нарушений: генные (моногенные - в основе патологии одна пара аллельных генов), хромосомные и мультифакториальные (болезни с наследственным предрасположением).
Моногенные заболевания — это нарушения, причиной которых являются мутации отдельных генов. Патологическое состояние у человека может быть вызвано изменением не только ядерных, но и митохондриальных генов. Большинство моногенных заболеваний наследуются в соответствии с законами Менделя, это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена. В группе моногенных заболеваний выделяют аутосомно-доминанантные, аутосомно-рецессивные, Х-сцепленные доминантные, Х-сцепленные рецессивные и митохондриальные патологические состояния.
Хромосомные синдромы сопровождаются аномалией количества или структуры хромосом у человека.
Мультифакториальные заболевания— это болезни с наследственной предрасположенностью, для проявления которых необходимо совместное действие наследственных и внешнесредовых факторов.
Причиной наследственной предрасположенности обычно является неблагоприятное сочетание в генотипе нескольких в принципе нормальных генов. Но патологические изменения в организме возникают только тогда, когда человек подвергается влиянию повреждающих факторов окружающей среды. Мультифакториальные заболевания разделяют в зависимости от того, какой орган поражен у больного человека. Например, выделяют заболевания нервной, сердечно-сосудистой, дыхательной, эндокринной систем и т.д.
Наследственные заболевания имеют свои особенности клинических проявлений, которые позволяют выделить их среди других патологических состояний у человека.
Семейный характер заболевания, когда имеются повторные случаи аналогичной патологии у членов одной семьи, прямо указывает на их возможную наследственность. Это особенно характерно для моногенных или мультифакториальных заболеваний. Однако хромосомные синдромы обычно выявляются только у одного человека в семье.
Множественные патологические изменения органов или систем. О наследственной причине заболевания позволяет думать первично вовлечение в патологический процесс многих органов или систем.
Это особенно характерно для моногенных и хромосомных синдромов. Вовлечение в процесс многих органов происходит в результате плейотропного эффекта, который дает большинство мутантных генов, вызывающих наследственные заболевания. Плейотропия – это, влияние одного гена на формирование нескольких признаков. Плейотропное действие гена является универсальной закономерностью, имеющей прямое отношение к наследственной патологии.
Контролирующие синтез коллагена и фибриллина мутантные аллели нескольких генов приводят к нарушению свойств соединительной ткани, которая является основой всех органов и тканей, становятся понятными множественные влияния этих мутаций на клинику наследственных болезней соединительной ткани, как, например, синдромы Марфана, Элерса-Данло: подвывих хрусталика, нарушение стенки аорты, пролапс митрального клапана и др.
Хроническое, рецидивирующее течение заболевания приводит к постепенному ухудшению клинической картины и состояния больного. Например, дети с миодистрофией Дюшенна постепенно теряют двигательную активность из-за атрофии мышц. Патологический процесс очень трудно, а подчас и невозможно приостановить. Хронический процесс при наследственной патологии развивается в результате постоянного действия мутантного гена. Рецидивирующее течение наследственных болезней вызвано и генетическим действием, и действием внешних факторов. В результате наследственные заболевания с большой степенью вероятности могут сопровождаться инвалидностью и ухудшением состояния больного.
Врожденный характер заболевания. Не менее 25% всех форм генных наследственных болезней и почти все хромосомные заболевания начинают формироваться внутриутробно. Если ребенок рождается с комплексом патологических признаков, то болезнь можно считать врожденной (например, ихтиоз, аутосомная микроцефалия и др.). Большинство пороков развития и хромосомных синдромов обнаруживаются уже при рождении ребенка. Однако многие моногенные мультифакториальные заболевания начинают развиваться значительно позже, иногда только в старческом возрасте. В то же время врожденная патология не всегда является наследственной.
Примером врожденных, но не наследственных болезней являются талидомидный, сифилитический, алкогольный синдромы, причина возникновения которых устанавливается при сборе анамнеза, относящегося к первым неделям беременности. Так, некоторые пороки развития, обнаруживаемые у новорожденного ребенка, формируются под влиянием повреждающих факторов внешней среды, действовавших на плод во время беременности.
Специфические симптомы наследственных болезней или их сочетания дают основание думать о наследственной природе заболевания. Например, голубой цвет склеры, который не влияет на функцию органа зрения, бывает при несовершенном остогенезе и некоторых других болезнях соединительной ткани. От больных фенилкетонурией исходит мышиный запах. При кровоточивости можно думать о гемофилии или болезни фон Виллебранта. Больные с мукополисахаридозами имеют грубые черты лица. При ахондроплазии выражены непропорциональность конечностей и туловища, низкий своеобразный лицевой череп. Астеническое телосложение с деформированной грудной клеткой встречается при синдроме Марфана. Клиническая диагностика наследственных болезней опирается только на определенные патологические признаки у больного или их специфическое сочетание, но и на особенности строения частей тела (например: форма лба, носа, разрез глаз, граница роста волос которые не нарушают функции организма. Так, при несовершенном остеогенезе у одного человека можно обнаружить частые переломы и деформации костей конечностей, тугоухость и голубой цвет белков глаз, который не влияет на функцию органа зрения.
Устойчивость к наиболее распространенным методам терапии. Наследственные болезни имеют особенность – устойчивость к лечению. До настоящего времени лечение многих наследственных заболеваний, оказывается, очень сложным, потому что исправить первичные звенья не всегда удается (нейрофиброматоз, миодистрофия, Дюшенна). Лечение поражения бронхов у больного муковисцидозом требует постоянного приема современных сильнодействующих лекарственных препаратов, использование которых, однако, не приводит к выздоровлению пациента. Но не все болезни устойчивы к терапии. При ключевых звеньев патогенеза разрабатываются лечения.
Хромосомные болезни
Хромосомный комплекс нормальных соматических клеток современного человека состоит из 46 хромосом (2п=46). В клетка, индивидуума женского пола кроме 44 аутосом имеется пара половых хромосом ХХ, а у лиц мужского пола - ХУ. Принятые формулы для изображения соответствующих кариотипов представлены следующим образом: 46, ХХ; 46, ХУ.
Хромосомные болезни — это большая группа врожденных патологических состояний с множественными врожденными пороками развития, причиной которых является изменение количества или структуры хромосом. Возникают они в результате мутаций в половых клетках одного из родителей. Тогда все клетки организма больного имеют аномальный кариотип. Из поколения в поколение передаются не более 3-5% из них.
Однако изменение набора хромосом может произойти и во время первых делений зиготы, даже если она была сформирована из нормальных гамет. В таких случаях образуется мозаицизм, т.е. часть снеток организма имеет другой набор хромосом.
Клиническое описание этих заболеваний появилось еще до открытия их хромосомной природы. Наиболее часто встречающаяся болезнь, трисомия 21, особый вид умственной отсталости у детей, была описана и 1866 г. английским педиатром Дауном и получила название «синдрома Дауна». Н.А. Шерешевским в 1925 г. дано первое клиническое описание синдрома моносомии по Х-хромосоме, а затем Г. Тернер и 1938 г. описал этот синдром. По фамилии этих ученых миосомию называют синдромом Шерешевского—Тернера. Г. Клайнфельтер впервые описал аномалии в системе половых хромосом у мужчин — трисомия ХХУ. Изучение хромосомных болезней активно началось с 60-х гг. XX в. благодаря широкому развертыванию цитогенетических исследований, когда полностью сложилась клиническая цитогенетика. Учение о хромосомной патологии сложилось в результате интенсивного изучения хромосом человека и хромосомных болезней.
Была показана роль хромосомных и геномных мутаций в патологии человека, расшифрована хромосомная причина многих синдромов врожденных пороков развития. В настоящее время описано около
1000 различных видов аномалий хромосом у человека. Примерно 100 форм имеют клинически очерченную картину и называются синдромами.
Распространенность хромосомных болезней одинакова во всех национальных и этнических группах и составляет в среднем 7-8 больных на каждую 1000 новорожденных. В России эта патология регистрируется примерно у 12 000 новорожденных ежегодно.
Хромосомные аберрации у плода часто являются причиной неблагополучного исхода беременности. Около 90% эмбрионов человека, имеющих эти аномалии, погибают еще внутриутробно. Примерно 50% всех диагностированных самопроизвольных абортов обусловлены хромосомными нарушениями. Подобные нарушения выявляются у 7% мертворожденных. Примерно 45% всех случаев множественных врожденных пороков у детей составляют хромосомные синдромы.
Различают геномные синдромы и структурные изменения хромосом. Геномные синдромы характеризуются изменением числа хромосом. У человека обнаружены только три типа геномных мутаций тетраплоидия, триплоидия и анеуплоидия.
Полиплоидия редко обнаруживается у новорожденных, у которых зарегистрированы случаи триплоидии (69 хромосом) и тетраплоидип (92 хромосомы). Это нарушение хромосомного набора чаще выявляется у эмбрионов при выкидышах в первом триместре беременности.
Анеуплоидия — это увеличение или уменьшение числа хромосом, некратное гаплоидному. Чаще всего у человека регистрируется наличие дополнительной хромосомы — трисомия по аутосомам. При этом какая-либо хромосома представлена в организме тремя копия ми, а кариотип включает 47 хромосом. Возможно и больше копий (4 или 5) одной хромосомы в организме. Увеличиваться может число как аутосом, так и половых хромосом. Отсутствие одной хромосомы называется моносомией. Встречаются полисомии по половым хромо сомам — три-, тетра-, пентасомии. Кариотип человека в этом случае содержит 45 хромосом. Совместимой с жизнью является только моносомия по Х-хромосоме (синдром Шерешевского—Тернера).
Все хромосомные болезни принято делить на две группы:
-
Связанные с аномалиями числа хромосом. В эту группу входи I три подгруппы:
-
болезни, причиной которых является нарушение числа хромосом.
-
болезни, связанные с увеличением или уменьшением числа половых X- и У-хромосом.
—- болезни, обусловленные полиплоидией, — кратным увеличением гаплоидного набора хромосом.
-
Связанные со структурными нарушениями (аберрациями) хромосом.
Их причинами являются:
-
Транслокации — обменные перестройки между негомологичными хромосомами.
-
Делеции — потери участка хромосомы.
-
Инверсии — повороты участка хромосомы на 180°.
-
Дупликации — удвоения участка хромосомы.
-
Изохромосомия — хромосомы с повторяющимся генетическим материалом в обоих плечах.
-
Возникновение кольцевых хромосом — соединение двух концевых делеций в обоих плечах хромосомы.
В настоящее время у человека известно более 700 таких заболеваний, вызванных структурными нарушениями хромосом. 25% приходится на аутосомные трисомии, 46% — на патологию половых хромосом. Среди хромосомных перестроек наиболее часто встречаются транслокации и делеции (рисунок 1).
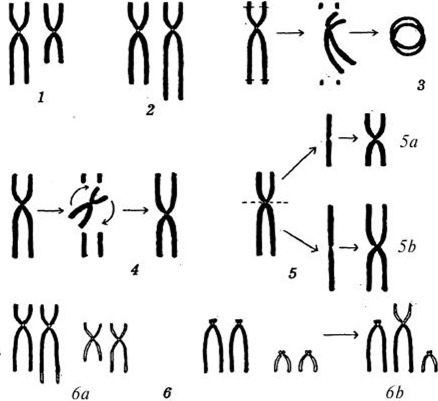
Рисунок 1- Основные типы структурных изменений хромосом:
1-делеция; 2 – дуплицкация; 3 – кольцевая хромосома; 4 –инверсия; 5а и 5в – изохромосомы; 6а и 6в - транслокации
Кроме классификации хромосомных болезней на геномные синдромы и структурные аберрации выделяют сбалансированные и несбалансированные нарушения. Если транслокация взаимная без потери участков, вовлеченных в нее хромосом, то она называется сбалансированной. Сбалансированные хромосомные аномалии обычно не сопровождаются фенотипическими нарушениями. При этом количество наследственного материала остается в норме, хотя структура хромосом изменяется. Сбалансированными аберрациями могут быть инверсии и транслокации.
Несбалансированные аномалии хромосом характеризуются утерей или удвоением части, или целой хромосомы. В таких ситуациях происходит значительное изменение состава генетического материала, что отражается на состоянии больного. 95% всех случаев несбалансированных нарушений — это геномные синдромы (из них 75 % — это синдром Дауна). Остальные 5% патологических состояний вызваны структурными аберрациями хромосом.
Здоровый человек имеет повышенный риск рождения ребенка с хромосомным синдромом. Причиной такого риска является случайное расхождение хромосом в процессе мейоза, что может привести к нарушению баланса генетического материала в гаметах. Если такая половая клетка вступит в оплодотворение, то может родиться больной ребенок.
Хромосомные синдромы обычно являются спорадическими, т.е. в семье регистрируется только один такой больной. Причиной и ого является то, что большинство изменений хромосом происходит и результате случайной мутации. Такие мутации чаще происходят при оогенезе, чем при сперматогенезе. Риск возникновения геномных мутаций в гаметах значительно увеличивается с возрастом женщины, особенно если она старше 35 лет.
Точно охарактеризовать хромосомную патологию и ее варианты позволяют три основных принципа:
-
Тип мутации — характеристика хромосомной или геномной мутации с учетом конкретной хромосомы (триплоидия, простая трисомия по хромосоме 21, частичная моносомия и т.д.).
-
Определение типа клеток, в которых возникла мутация, — в гаметах или зиготе. Мутации в гаметах ведут к полным формам хромосомных болезней. У таких индивидов все клетки несут унаследованную с гаметой хромосомную аномалию. Если хромосомная аномалия возникает в зиготе или на ранней стадии дробления, то развивается организм с клетками хромосомной конституции. Такие формы хромосомных болезней называются мозаичными.
-
Выявление поколения, в котором возникла мутация. Возникла она заново в гаметах здоровых родителей, спорадически или родители уже имели такую аномалию.
Степень тяжести физических и функциональных отклонений в организме больного определяется характером изменений хромосом. Например, геномные мутации приводят к более тяжелым нарушениям состояния здоровья ребенка, чем структурные аберрации. Мозаичные варианты трисомий сопровождаются менее выраженными изменениями фенотипа.
Данные о механизмах нарушений при хромосомных болезнях показывают, что можно выделить три типа генетических эффектом: специфические, связанные с изменением числа структурных геном, кодирующих синтез белка — при трисомии их число увеличивается, при моносомии уменьшается; полуспецифические эффекты, обусловленные изменением числа генов и в норме представленные в виде многочисленных копий. К таким генам относятся гены рибосомных и транспортных РНК, рибосомных и гистоновых белков, сократи тельных белков актина и тубулина. Эти белки контролируют этапы метаболизма клеток, процессов ее деления, межклеточных взаимодействий; неспецифические — связывают с измененным содержанием гетерохроматина в клетке.
Общим для всех форм хромосомных болезней является множественность поражения. Для них характерны врожденные пороки развития внутренних и наружных органов, замедленные внутриутробные и постнатальные рост и развитие, черепно-лицевые дизморфии, отставание психического развития, нарушения функций нервной, эндокринной и иммунной систем.
Степень отклонений в развитии организма зависит от качественной и количественной характеристики унаследованной хромосомной болезни. Нарушения нервно-психического и физического развития особенно характерны для изменений, затрагивающих количество или структуру аутосомных хромосом. Среди них чаще всего встречаются трисомии, в первую очередь — 21-й хромосомы. Значительно реже у новорожденных выявляются трисомии других хромосом. Большинство случаев подобных изменений хромосом обычно несовместимы с жизнью, даже при внутриутробном развитии плода. Так, трисомии составляют около 50% всех случаев хромосомных аберраций у эмбрионов при самопроизвольном прерывании беременности.
Моносомии аутосом не встречаются у новорожденных и крайне редко обнаруживаются при абортах. В экспериментах на млекопитающих было установлено, что отсутствие целой аутосомы приводит к гибели зародыша в первые дни после оплодотворения. Полные трисомии у живорожденных наблюдаются только по аутосомам, богатым гетерохроматином.
Частичные трисомии и моносомии обычно не сопровождаются столь грубыми нарушениями в жизнедеятельности организма и могут выявляться живорожденных детей. Подобные изменения в настоящее время описаны для всех аутосом.
Сбалансированные перестройки аутосом, как правило, не приводят к фенотипическим нарушениям. Однако в потомстве носителей таких аномалий повышена вероятность возникновения частичных моносомии или трисомии, т.е. несбалансированных изменений хромосом. Наиболее специфичные для того или иного синдрома проявления обусловлены отклонениями в содержании сравнительно небольших сегментов хромосом. Например, специфические клинические симптомы синдрома Дауна проявляются при трисомии по сегменту длинного плеча хромосомы 21q 22.1. Характерные черты синдрома Эдвардса связаны с трисомией сегмента хромосомы 18qll.
Каждой хромосомной патологии свойственен клинический полиморфизм. Проявления патологии могут быть очень широкими — от незначительных отклонений до летального эффекта. 60-70% случаев трисомии 21 заканчиваются гибелью во внутриутробном периоде, в 30% случаев рождаются дети с синдромом Дауна. Моносомия по X-хромосоме среди новорожденных — синдром Шерешевского—Тернера — это 10% всех моносомных по Х-хромосоме зародышей, остальные погибают. Если учесть еще доимплантационную гибель 1Игот Х0, то живорожденные с синдромом Шерешевского—Тернера составляют 1%.
Синдромы наиболее распространенных хромосомных болезней.
Синдром Дауна (трисомия 21) — часто встречающееся и хорошо изученное хромосомное заболевание. Впервые оно было описано м 1866 г. английским педиатром А. Дауном. В 1959 г. с соавторами установили, что причиной синдрома Дауна является наличие дополнительной 21-й хромосомы в кариотипе больного (рисунок 2).
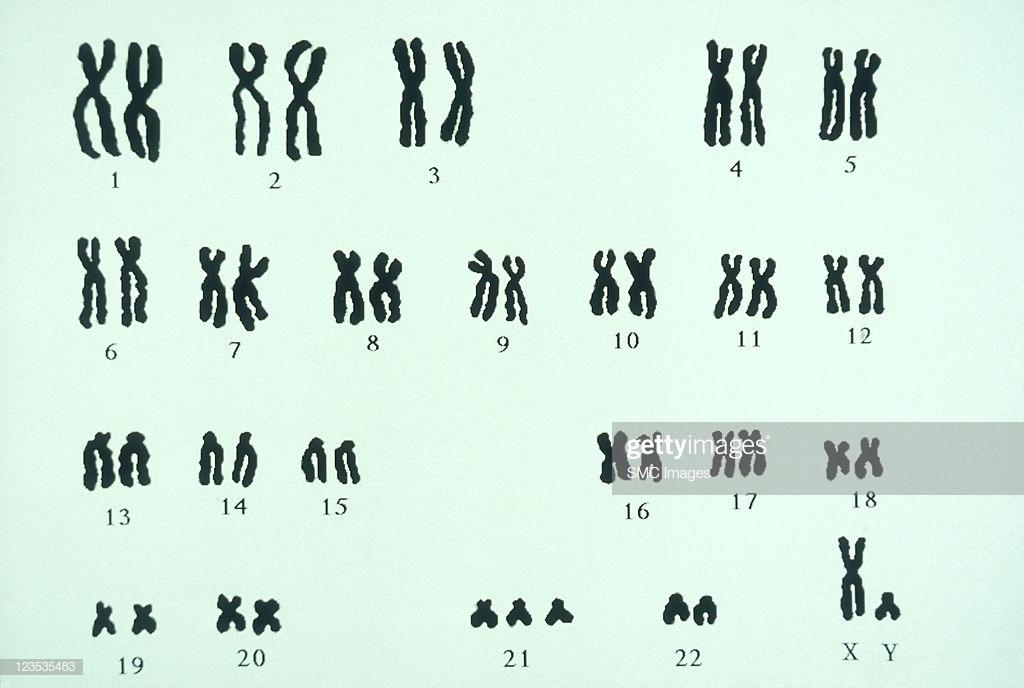
Рисунок 2 – Кариотип мальчика с синдромом Дауна (трисомия 21-й хромосомы)
Встречается у новорожденных с частотой 1 больной на 700-800 новорожденных. Частота рождения детей с болезнью Дауна зависит от возраста матери, женщин до 18 лет и старше 35 лет. 80-90% всех случаев заболевания являются результатом не расхождения хромосом в мейозе у матери. Но остальные 10-20% трисомий 21-й хромосомы вызваны нарушениями сперматогенеза у отца больного. Риск рождения больного ребенка у 40-45-летних матерей в 16 раз выше, чем в 20-24 года, и составляет 3%. Высокая частота рождения детей с синдромом Дауна наблюдается у матерей до 18 лет.
Синдром Дауна может возникнуть в результате разных вариантов изменений хромосом, приводящих к увеличению числа 21-й хромосомы.
-
Основную долю (94%) составляют случаи простой полной трисомии 21-й хромосомы. Кариотип таких больных можно записать следующим образом: 47, ХУ,+21 или 47, XX,+21.
-
Около 2% больных детей имеют мозаичные варианты синдрома Дауна. При этом часть клеток организма больного содержит дополнительную хромосому, а другие клетки имеют нормальный кариотип. Мозаичные варианты синдрома Дауна обычно сопровождаются менее выраженными изменениями фенотипа по сравнению с простой трисомией.
-
Почти 50% транслокационных форм наследуются от родителей-носителей, а 50% транслокаций возникают вновь. Иногда заболевание возникает при транслокации участка длинного плеча 21-й хромосомы. Эта форма заболевания регистрируется примерно в 4% случаев.
Клинические проявления синдрома Дауна разнообразны и заметны уже при рождении.
При беременности характерны: токсикоз и угроза выкидыша. Для синдрома Дауна характерны: форма головы с уплощенным затылком; толстая кожная складка на задней поверхности шеи; скошенный и узкий лоб; лицо плоское, переносица широкая и вдавленная; язык у больных большой и виден между губами; постоянно открытый рот, толстые губы; «монголоидный» разрез глаз; типичен эпикант; ушные раковины уменьшены и деформированы. Со стороны костномышечной системы характерны: низкий рост; короткая шея; воронкообразная или килевидная грудина; широкие кисти и стопы с короткими пальцами; глубокие борозды на ладонях; первый палец на стопах широко отстоит от других пальцев — «сандалевидный промежуток»; мышечная гипотония с разболтанностью суставов. Поэтому новорожденные дети с синдромом Дауна обычно лежат в кроватке, раскинув ручки и ножки.
При синдроме Дауна около 50% больных имеют врожденные пороки сердца. Обычно это дефекты межпредсердной или межжелудочковой перегородок. Пищеварительный тракт поражается в 15% случаев, в первую очередь, атрезии или стенозы двенадцатиперстной кишки. Возможны также нарушения формирования и других органов.
Для всех больных этим синдромом характерна умственная отсталость — дебильность — в 75% случаев, имбецильность — у 20% больных, идиотия — в 5% случаев. При синдроме Дауна отмечается задержка физического и умственного развития, формирования моторных навыков и речи. Дети позже начинают ходить и говорить. У них резко нарушено абстрактное мышление. Они легче осваивают навыки, связанные с физическими движениями, чем речевые. Дети с синдромом Дауна внимательные, ласковые, послушные и общительные, терпеливые при обучении.
Продолжительность жизни при синдроме Дауна короче, чем у здоровых людей. Врожденные пороки внутренних органов, сниженная приспособляемость детей с синдромом Дауна часто приводят к смерти в первые 5 лет. Такие дети значительно чаще страдают острыми инфекциями и злокачественными заболеваниями крови. Средняя продолжительность жизни больных составляет 20 лет. Примерно треть детей с синдромом Дауна погибают на первом году жизни. Очень редко больные доживают до 50 лет.
Лечение больных с синдромом Дауна должно быть комплексным и неспецифичным:
1) Развитие моторных навыков и всех органов чувств: зрения, слуха, осязания, обоняния.
2) Полноценное питание, массаж и гимнастика, развивающие занятия.
3) Использование ноотропных лекарственных средств, препаратов, укрепляющих ЦНС лекарственной терапии.
4) Стимуляции двигательной активности ребенка — в возрасте от 0 до 2 месяцев необходимо в течение дня несколько раз поворачивать на животик, при этом под грудь подкладывают небольшую подушечку. В возрасте от 2 до 6 месяцев необходимо переворачивать ребенка на бочок и на животик.
-
В возрасте от 6 до 12 месяцев в процессе занятий необходимо обучать пациента присаживаться и самостоятельно сидеть.
Многие больные с трисомией 21 способны жить самостоятельно, создавать семьи, овладевать несложными профессиями. С помощью специальных методов обучения, укрепления здоровья, правильного питания и ухода, проведения необходимого лечения можно таким больным продлить жизнь.
Синдром Патау (трисомия по 13-й хромосоме) был впервые описан в 1960 г. К. Патаи с соавторами у детей с множественными пороками развития. Синдром Патау оказался вторым патологическим состоянием человека, при котором были установлены изменения хромосом. Это заболевание встречается у детей с частотой и среднем 1 больной на 6 000 новорожденных. Мальчики и девочки поражаются одинаково часто. Часть детей с синдромом Патау погибают в пренатальном периоде.
80-85% всех случаев заболевания обусловлены не расхождением хромосом в мейозе, процессе формирования половых клеток родителей, т.е. это является результатом спонтанной мутации. Вероятность возникновения таких мутаций увеличивается с возрастом матери, как и при синдроме Дауна. Кариотип таких больных: 47, XX, + 13 или 47, ХУ, + 13.
Примерно 15% случаев синдрома Патау являются результатом транслокации 13-й хромосомы на какую-нибудь из хромосом группы Б. Очень редко встречаются другие цитологические случаи этого синдрома: мозаицизм, изохромосомы, другие транслокации и т.д.
Хромосома 13 значительно крупнее 21-й хромосомы, и соответственно ее трисомия вызывает значительно более тяжелые структурные и функциональные нарушения в организме ребенка. Для беременности таким плодом характерны в 50% случаев многоводие и угроза выкидыша. Роды наступают несколько раньше обычного срока — на 36-38-й неделе. Масса тела новорожденного с синдромом Патау ниже, чем должна быть при соответствующем сроке беременности.
Синдром Патау сопровождается множественными врожденны ми пороками развития головного мозга и лица. Окружность черепа уменьшена, что приводит к формированию микроцефалии. В теменной области волосистой части головы часто выявляется участок отсутствия кожи до 1 см в диаметре. Лоб скошенный, глазные щели узкие, переносица запавшая, глаза недоразвиты (микрофтальмия) с помутнением роговицы, ушные раковины расположены низко и деформированы. Типичным признаком синдрома Патау у всех больных являются расщелины верхней губы и неба, часто двухсторонние.
Всегда обнаруживаются пороки нескольких наружных и внутренних органов: полидактилия на руках и чаще двухсторонняя, флексорное положение кистей, дефекты перегородок сердца, незавершенный поворот кишечника, кисты почек, аномалии внутренних половых органов, дефекты поджелудочной железы и печени. Пороки почек при синдроме Патау встречаются у большинства больных. Почки увеличены в размерах, содержат много кист в корковом слое. Типичными для детей с трисомией 13-й хромосомы являются пороки развития половых органов: неопущение яичек (крипторхизм) и недоразвитие полового члена у мальчиков, удвоение матки и влагалища у девочек. При подозрении на синдром Патау показано УЗИ всех внутренних органов.
В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау живут недолго, и 95% таких больных умирают до года жизни. Однако некоторые больные с синдромом Патау живут несколько лет. У таких больных выражена задержка психомоторного развития, имеется идиотия.
Лечение детей с синдромом Патау неспецифическое. Проводятся операции по поводу врожденных пороков развития, общеукрепляющее лечение, профилактика инфекционных и простудных заболеваний. Тщательный уход за такими пациентами облегчает их состояние, предупреждает инфекционные осложнения. Количество и состав основных ингредиентов пищи для таких больных должны соответствовать их возрасту. Но часто необходимо увеличить число кормлений с уменьшением разовой дозы, так как дети быстро устают. После кормления ребенку дают выпить несколько миллилитров кипяченой йоды, чтобы во рту не оставалось частиц пищи. Необходимо также постоянно контролировать стул и мочеиспускание у ребенка, своевременно проводить туалет промежности и смену пеленок. Дети с синдромом Патау в основном глубокие идиоты.
Синдром Эдвардса (трисомия 18) был описан в 1960 г. Почти во всех случаях синдром Эдвардса обусловлен регулярной трисомией 18-й хромосомы. Кариотип больных при этом заболевании 47, XX, + 18 или 47, ХУ, + 18.
Мозаицизм и транслокационные формы встречаются редко. Частота больных среди новорожденных — 1 больной ребенок на 7 000 новорожденных. Соотношение девочек и мальчиков с синдромом Эдвардса составляет 1:3.
Беременность при синдроме Эдвардса также осложняется угрозе прерывания и многоводием. Весьма характерно несоответствие размеров плода сроку беременности. При рождении дети имеют очень низкую массу тела, в среднем 2170 г при доношенной беременности при синдроме Эдвардса отмечается выраженная задержка пренатального развития при нормальной продолжительности беременности.
Синдром Эдвардса — это множественные врожденные пороки развития лицевой части головы, сердца, костной системы, половых органов. Чаще всего регистрируются дефекты развития конечностей, недоразвитие больших пальцев рук и лучевых костей, неправильно сформированные стопы с выступающей пяткой и провисанием свод;» (стопа-качалка), укорочение первой плюсневой кости. Иногда обнаруживаются спинномозговые грыжи и расщелины верхней губы, недоразвитие глаз — микрофтальмия.
Череп имеет долихоцефалический, выступающий затылок, глазные щели короткие, нижняя челюсть и отверстие рта маленькие, челюсть скошена назад, ушные раковины деформированы и расположены низко, слуховой проход узкий, грудина укорочена, грудная клетка широкая. Отмечается флексорное положение кистей рук. При этом III и IV пальцы прижаты к ладони и частично перекрыты II и V пальцами. Имеется аномальная стопа—пятка выступает, а свод стопы провисает, первый палец стоп короче второго пальца. Мышечный тонус у таких больных обычно повышен. Дети лежат в кроватке, отведя голову назад, с согнутыми конечностями. У большинства пациентов с синдромом Эдвардса определяются пороки сердца и чаще всего — дефект межжелудочковой перегородки. Этот порок может сочетаться с другими нарушениями строения сердца и крупных сосудов, например, с недоразвитием клапанов аорты и легочной артерии.
Артезия (отсутствие отверстия) пищевода, и т.д. У таких детей отмечаются недоразвитие (гипоплазия) легких, сращение почек, удвоение мочеточников, неопущение яичек (крипторхизм) у мальчиков.
Дети с синдромом Эдвардса умирают до года от осложнения, связанных с врожденными пороками развития, таких как асфиксия, сердечно-сосудистая недостаточность, пневмония, кишечная непроходимость. Для больных старшего возраста характерна глубоким умственная отсталость.
Уход за больными в основном заключается в предупреждении инфекционных осложнений. Больным с синдромом Эдвардса необходимо обеспечить полноценное питание, с увеличением частоты кормлений.
Синдромы с числовыми аномалиями половых хромосом.
Полисомии по половым хромосомам представляют собой большую группу хромосомных болезней с различными комбинациями дополнительных X- или У-хромосом и комбинациями разных клоном в случае мозаицизма. Общая частота полисомии по X- или У-хромосомам среди новорожденных составляет 1,5:1000-2:1000. В основном это полисомии XXX, ХХУ и ХУУ. 25% составляют мозаичные типы полисомий по половым хромосомам.
В отличие от аутосомных трисомий клиническая картина этих патологических состояний характеризуется нарушением полового развития и незначительным снижением интеллекта.
Синдромы полисомии Х-хромосомы включают в себя трисомию X (кариотип пациентки — 47, XXX), тетрасомию X (48, ХХХХ), пентасомию X (49, ХХХХХ). Дополнительные Х-хромосомы у таких людей инактивируются, поэтому такие патологические состояния являются совместимыми с жизнью, несмотря на выраженный хромосомный дисбаланс.
Трисомия Х-хромосомы среди новорожденных девочек составляет 1:1000. Обычно такая патология обнаруживается при случайном массовом обследовании детей. Женщины с кариотипом XXX в полном или мозаичном варианте имеют нормальное физическое и психическое развитие. Объясняется это тем, что в клетках две Х-хромосомы гетерохроматинизированы, а функционирует лишь одна, как у нор мальной женщины. Однако при трисомии X с возрастом увеличивается риск возникновения психических заболеваний. Интеллектуальное развитие нормальное или на нижней границе нормы. У некоторых женщин имеются нарушения репродуктивной функции — аменорея, дисменорея, ранняя менопауза. Редко обнаруживаются другие патологические изменения: микроцефалия, косоглазие, сколиоз, высокий рост, нарушение менструальной функции и бесплодие.
Увеличение числа Х-хромосом в кариотипе сопровождается усугублением поражения нервной системы, формированием пороков равития и нарушением функции половых органов. При тетрасомии и иентасомии Х-хромосомы у пациенток отмечаются значительная умственная отсталость, судороги, недоразвитие гениталий, пороки развития конечностей (их маленькие размеры, сращение лучевой и локтевой костей), врожденные пороки сердца, необычный внешний вид. Варианты синдрома Х-полисомии без У-хромосомы с числом более трех встречаются редко. С увеличением числа дополнительных \ хромосом нарастают отклонения от нормы. Диагностика полисомии \ хромосомы основывается на исследовании кариотипа. Заключение об избыточном количестве Х-хромосом можно предварительно сделать на основании исследования полового хроматина. Снижение интеллекта от пограничной умственной отсталости до различных (степеней олигофрении, черепно-лицевые дисформии, аномалии зубов, скелета и половых органов описаны у 2/3 больных женщин с тетра- и пентасомией.
Методика терапии при синдроме полисомии по Х-хромосоме диктуется теми патологическими нарушениями, которые отмечаются у пациенток.
Синдром Шерешевского—Тернера (45, X) — это единственная форма у рожденных детей, с кариотипом 45, X, не менее 90% зачатий абортируется самопроизвольно. Впервые этот синдром был описан русским ученым Н. А. Шерешевским в 1925 г. В 1938 г. Г. Тернер дал полное описание этого заболевания, которое получило название синдрома Шерешевского—Тернера. Позже была установлена этиология заболевания — моносомия по Х-хромосоме у женщин.
Частота этого синдрома составляет 1:3000 новорожденных девочек. Чаще всего при этом заболевании регистрируется полная моносомия по Х-хромосоме (60% всех больных) (рис. 9.10), из них м 80-85% случаев имеет материнское происхождение, а в 15-20% — отцовское.
Кариотип больных при этом заболевании включает 45 хромосом с одной Х-хромосомой (45, X). Кроме истинной моносомии во всех клетках встречаются другие формы хромосомных аномалий по половым хромосомам. Среди них делеции короткого или длинного плеча Х-хромосомы (46, X, Хр-; 46, X, Хq-). Часто выявляются формы мозаицизма, примерно в 20% всех случаев (рисунок 3).
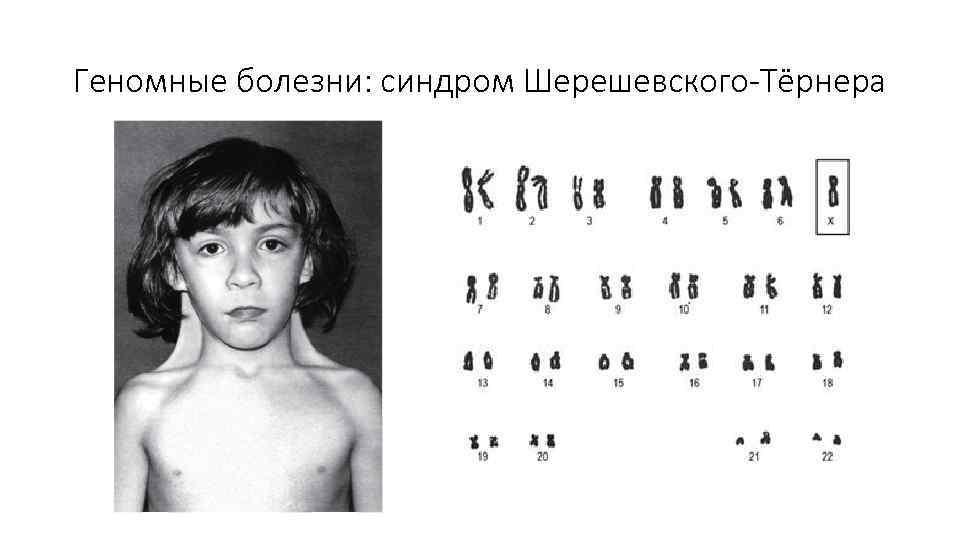
Рисунок 3 – Синдром Шерешевского-Тернера
Беременность плодом с этим заболеванием может также сопровождаться угрозой выкидыша, задержкой увеличения размеров плода Масса и длина тела новорожденных девочек с синдромом Шерешевского—Тернера несколько меньше, чем у здоровых детей.
Для синдрома Шерешевского—Тернера характерны: гипогонадизм, недоразвитие половых органов и вторичных половых призма ков; врожденные пороки развития; низкий рост. В первые месяцы жизни у ребенка проявлением заболевания являются короткая шеи с избытком кожи и крыловидными складками, лимфатический отек на тыльных поверхностях кистей рук, предплечий, стоп и голеней. Подобные изменения развиваются не у всех девочек с синдромом Шерешевского—Тернера. Часто в первые годы жизни у детей встречаются разные врожденные пороки сердца и почек, до 20% случаев.
В подростковом возрасте можно заметить задержку роста ребенка. Необходимо учитывать, что 80% всех случаев низкорослости у девочек обусловлены синдромом Шерешевского—Тернера. Рост взрослых девушек с этим заболеванием составляет в среднем 135-140 см. Характерным для синдрома Шерешевского—Тернера является задержка полового развития у девочек 12-14 лет.
Телосложение у таких пациентов коренастое. На коже часто расположено множество родинок. Подбородок несколько выступает вниз, уши расположены низко. Шея широкая и короткая. Иногда по бокам ее расположены кожные складки — птеригиумы. Линия роста волос на задней поверхности шеи снижена. Грудная клетка широкая, «щитообразная». Часто выявляются различные деформации скелета: воронкообразная грудная клетка, вальгусное положение предплечий и голеней и др. Интеллектуальное развитие пациенток с этим заболеванием близко к норме. Однако для таких Пильных характерны инфантильность и снижение познавательных (способностей.
У взрослых с синдромом Шерешевского—Тернера отмечают нарушение скелета, черепно-лицевые дисформии, вальгусную девиацию коленных и локтевых суставов, остеопороз, бочкообразную грудную клетку, низкий рост волос на шее, птоз, эпикант, ретрогению, низкое расположение ушных раковин, бесплодие. Но было известно несколько случаев беременностей у женщин с полной моносомией по Х-хромосоме.
Мозаичные формы заболевания характеризуются стертыми клиническими проявлениями. Часть таких больных могут иметь нормальное или только немного задержанное половое развитие, регулярные менструации. Такие пациентки иногда имеют потомство.
Лечение при этом заболевании комплексное, направлено на исправление имеющихся аномалий. До 12-14 лет таким девочкам проводится терапия, корригирующая задержку роста. С 13-14 лет проводится гормональное лечение препаратами женских половых гормонов, которые вызывают формирование вторичных половых признаков и менструального цикла. При необходимости оперативно исправляются врожденные пороки развития или с помощью пласт и ческой хирургии — косметические нарушения. Современные методы гормональной терапии и экстракорпорального оплодотворения с использованием донорской яйцеклетки уже дали возможность рождении здорового ребенка нескольким женщинам, имеющим моносомию по Х-хромосоме. Проводится психотерапия. Своевременное применение всех методов лечения, применение генно-инженерного гормона роста обеспечивает почти полную компенсацию патологических проявлений у этих больных.
Синдром Клайнфельтера — наиболее часто встречающийся с типичной картиной синдром с набором 47, ХХУ. Включает случаи полисомии по половым хромосомам в кариотипе у мужчины при которых имеется не менее двух Х-хромосом и не менее одной У-хромосомы. Впервые был описан в 1942 г. Частот,1 синдрома Клайнфельтера в полном и мозаичном вариантах — 1 больной ребенок на 500-700 новорожденных. Около 80% всех случаев заболевания связаны с присутствием в кариотипе дополнительной Х-хромосомы (47, ХХУ). У таких мальчиков можно обнаружить тельца Барра в 15-25% эпителиальных клеток слизистой щек, барабанные палочки». У них также определяются и Р-тельца в 65% лимфоцитов крови. У других пациентов с синдромом Клайнфельтера в основном регистрируются мозаичные формы, когда, например, часть клеток организма больного может содержать нормальный набор хромосом.
Больные с синдромом Клайнфельтера высокого роста. Часто встречаются диспропорционально длинные нижние конечности, сколиоз, деформация грудной клетки. У некоторых больных с этим синдромом выявлялись катаракты, снижение слуха, врожденные пороки сердца, варикозное расширение вен. Достаточно часто у мальчиков с синдромом Клайнфельтера регистрируются умственная отсталость легкой степени или неустойчивость внимания, повышенная утомляемость, повышенная внушаемость, снижение инициативности, незрелость суждений. Степень тяжести умственной отсталости нарастает с увеличением числа Х-хромосом в кариотипе больного. Так, больные с 49, ХХХХУ страдают глубокой олигофренией. И то же время интеллект у людей с 47, ХХУ может быть нормальным.
В лечении таких пациентов используют препараты мужских половых гормонов, которые корректируют вторичные половые признаки.
Частота этой патологии составляет 1: 1 000 новорожденных мальчиков и возрастает до 10% у мужчин выше 2 м. Значительно реже регистрируются трисомии У-хромосомы (48, ХУУ У) и тетрасомии У-хромосомы (49, ХУУУУ). По физическому и умственному развитию они в большинство случаев не отличаются от нормальных индивидов. Заметных отклонений в половой и эндокринной системах нет. Такие мужчины могут иметь детей. Однако для 35% пациентов с полисемией У-хромосомы характерны ускорение роста в детском возрасте, высокий рост у взрослого — не ниже 186 см, удлинение конечностей, грубые черты лица, выступающие надбровные дуги и переносье, увеличенная нижняя челюсть, большие ушные раковины, патология коленных и локтевых суставов, нарушение поведения (агрессивность, асоциальные поступки). У некоторых больных с возрастом развиваются шизофрения и эпилепсия. Часто регистрируется умственная отсталость, степень тяжести которой также зависит от количества У-хромосом в карио типе мальчика. Чем их больше, тем значительней интеллектуальная недостаточность.
Лечение при дисомии по У-хромосом чаще не требуется. При наличии показаний проводятся гормонотерапия при недоразвитии гениталий, противосудорожная терапия и т.д.
Заболевания, обусловленные внутрихромосомными перестройками.
К синдромам, обусловленным внутрихромосомными перестройками, относятся частичные трисомии и моносомии аутосом наряду с делециями, инверсиями и дупликациями. Структурные аномалии хромосом обычно сопровождаются меньшим генным дисбалансом, чем полные трисомии, поэтому они описаны у живорожденных детей для всех типов аутосом.
Синдром «кошачьего крика» (5р-) — это частичная моносомия по короткому плечу хромосомы 5 (5р-). Заболевание было описано в 1963 г. Дж. Леженом. У детей с умственной отсталостью и необычным лицом было обнаружено уменьшение размера короткого плеча хромосомы из группы В. Причиной данного заболевания является делеция от 1/3 1/2 длины короткого плеча одной из 5-й пары хромосом (рис. 9.14). Соотношение мужчин и женщин составляет 1:1,3. Кариотип больного ребенка 46, XX, 5р- или 46, ХУ, 5р-. Синдром моносомии 5р- является самым частым среди заболеваний, вызванных структурным изменением хромосом. Частота синдрома достаточно высокая для структурных синдромов — на 1 новорожденного из 45 000.
У девочек данный синдром регистрируется несколько чаще. Клиническая картина синдрома моносомии 5р характеризуется необычным плачем, напоминающим требовательный кошачий крик или мяуканье, обусловленным сужением гортани, мягкостью хрящей, меньшими размерами надгортанника, необычной складчатостью слизистой оболочки. По этой причине этот синдром был назван синдромом «кошачьего крика».
Основная часть всех случаев заболевания связана с утратой части короткого плеча 5-й хромосомы. Вероятность рождения детей с синдромом «кошачьего крика» не зависит от возраста родителей. Беременность часто сопровождается угрозой выкидыша. Масса тела такого новорожденного ребенка обычно несколько ниже нормальной.
Некоторые больные с синдромом 5р- достигают зрелого возраста, хотя все же большинство из них погибает в раннем детстве от пневмонии или сердечно-сосудистой недостаточности.
Клиническая картина синдрома 5р у грудного ребенка характеризуется специфическим криком, напоминающим кошачий крик, обусловленный сужением гортани или патологией голосовых связок.
Почти у всех больных имеются различные изменения мозговой части лица и черепа. У больных отмечаются лунообразное лицо, гипертелоризм, микроцефалия, микрогения, эпикант, высокое нёбо, плоская спинка носа. Наружные уголки глаза располагаются ниже внутренних (антимонголоидный разрез). Мышечный тонус несколько снижен. С увеличением возраста ребенка специфический крик и мышечная гипотония постепенно исчезают. Кроме того, встречаются врожденные пороки сердца и некоторых других внутренних органов.
Вместе с тем становятся все более заметными задержка умственного и физического развития, микроцефалия (малые размеры черепа), обнаруживаются косоглазие, атрофия зрительных нервов, изменение сетчатки.
Продолжительность жизни больных с синдромом 5р- зависит от тяжести врожденных пороков внутренних органов, медицинской помощи. Большинство больных умирают в первые годы жизни, некоторые достигают 10-летнего возраста. Единичные больные достигают 50-летнего возраста.
Приложение 1
1.Что такое наследственность?
2.Что такое хромосома?
3.Что такое патология?
Эталоны ответов:
1. Наследственность — способность организмов передавать свои признаки и особенности развития потомству из поколения в поколение.
2. Хромосома — главные структурно-функциональные элементы клеточного ядра, содержащие расположенные в линейном порядке гены и обеспечивающие хранение, воспроизводство генетической информации, а также начальные этапы ее реализации в признаки
3. Патология — раздел медицинской науки, изучающий болезненные процессы и состояния в живом организме.
Приложение 2
Обобщение и систематизация знаний:
1.Какова классификация наследственных заболеваний?
2.Дайте определение понятию хромосомная болезнь
3.Перечислите хромосомные заболевания человека, как их можно классифицировать?
4.Что вы знаете о врожденной патологии?
5.Дайте характеристику синдрома Дауна, синдром Эдвардса, синдром Патау, Шерешевского-Тернера, Синдром Клайнфельтера, синдром Кошачьего Крика.
Эталоны ответов
1. Все наследственные болезни делятся по характеру изменения наследственных структур на три основных типа нарушений: генные (моногенные - в основе патологии одна пара аллельных генов), хромосомные и мультифакториальные.
Моногенные заболевания — это нарушения, причиной которых являются мутации отдельных генов. Патологическое состояние у человека может быть вызвано изменением не только ядерных, но и митохондриальных генов. Большинство моногенных заболеваний наследуются в соответствии с законами Менделя, это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена.
Хромосомные синдромы сопровождаются аномалией количества
или структуры хромосом у человека.
Мультифакториальные заболевания — это болезни с наследственной предрасположенностью, для проявления которых необходимо совместное действие наследственных и внешнесредовых факторов.
2. Хромосомные болезни — это большая группа врожденных патологических состояний с множественными врожденными пороками развития, причиной которых является изменение количества или структуры хромосом. Возникают они в результате мутаций в половых клетках одного из родителей. Тогда все клетки организма больного имеют аномальный кариотип. Из поколения в поколение передаются не более 3-5% из них.
3. Все хромосомные болезни принято делить на две группы:
Связанные с аномалиями числа хромосом. В эту группу входит три подгруппы:
-болезни, причиной которых является нарушение числа хромосом.
-болезни, связанные с увеличением или уменьшением числа половых X- и У-хромосом.
-болезни, обусловленные полиплоидией, — кратным увеличением гаплоидного набора хромосом.
Связанные со структурными нарушениями (аберрациями) хромосом.
-Их причинами являются:
-Транслокации — обменные перестройки между негомологичными хромосомами.
-Делеции — потери участка хромосомы.
-Инверсии — повороты участка хромосомы на 180°.
-Дупликации — удвоения участка хромосомы.
-Изохромосомия — хромосомы с повторяющимся генетическим материалом в обоих плечах.
-Возникновение кольцевых хромосом — соединение двух концевых делеций в обоих плечах хромосомы.
4. Почти все хромосомные заболевания начинают формироваться внутриутробно. Если ребенок рождается с комплексом патологических признаков, то болезнь можно считать врожденной (например, ихтиоз, аутосомная микроцефалия и др.). Большинство пороков развития и хромосомных синдромов обнаруживаются уже при рождении ребенка. Однако многие моногенные мультифакториальные заболевания начинают развиваться значительно позже, иногда только в старческом возрасте. В то же время врожденная патология не всегда является наследственной.
5. Синдром Дауна (трисомия 21) — часто встречающееся и хорошо изученное хромосомное заболевание. Впервые оно было описано в 1866 г. английским педиатром А. Дауном. Причиной синдрома Дауна является наличие дополнительной 21-й хромосомы в кариотипе больного.
Для синдрома Дауна характерны: форма головы с уплощенным затылком; толстая кожная складка на задней поверхности шеи; скошенный и узкий лоб; лицо плоское, переносица широкая и вдавленная; язык у больных большой и виден между губами; постоянно открытый рот, толстые губы; «монголоидный» разрез глаз; типичен эпикант; ушные раковины уменьшены и деформированы. Со стороны костномышечной системы характерны: низкий рост; короткая шея; воронкообразная или килевидная грудина; широкие кисти и стопы
с короткими пальцами; глубокие борозды на ладонях; первый палец на стопах широко отстоит от других пальцев — «сандалевидный промежуток»; мышечная гипотония с разболтанностью суставов. Поэтому новорожденные дети с синдромом Дауна обычно лежат в кроватке, раскинув ручки и ножки.
При синдроме Дауна около 50% больных имеют врожденные пороки сердца.
Синдром Эдвардса обусловлен регулярной трисомией 18-й хромосомы. Кариотип больных при этом заболевании 47, XX, + 18 или 47, ХУ, + 18.
Беременность при синдроме Эдвардса также осложняется угрозе прерывания и многоводием. Весьма характерно несоответствие размеров плода сроку беременности. При рождении дети имеют очень низкую массу тела, в среднем 2170 г при доношенной беременности при синдроме Эдвардса отмечается выраженная задержка пренатального развития при нормальной продолжительности беременности.
Синдром Эдвардса — это множественные врожденные пороки развития лицевой части головы, сердца, костной системы, половых органов. Чаще всего регистрируются дефекты развития конечностей, недоразвитие больших пальцев рук и лучевых костей, неправильно сформированные стопы с выступающей пяткой и провисанием свод;» (стопа-качалка), укорочение первой плюсневой кости. Иногда обнаруживаются спинномозговые грыжи и расщелины верхней губы, недоразвитие глаз — микрофтальмия.
Для синдрома Шерешевского—Тернера характерны: гипогонадизм, недоразвитие половых органов и вторичных половых призма ков; врожденные пороки развития; низкий рост. В первые месяцы жизни у ребенка проявлением заболевания являются короткая шеи с избытком кожи и крыловидными складками, лимфатический отек на тыльных поверхностях кистей рук, предплечий, стоп и голеней. Подобные изменения развиваются не у всех девочек с синдромом Шерешевского—Тернера. Часто в первые годы жизни у детей встречаются разные врожденные пороки сердца и почек, до 20% случаев.
В подростковом возрасте можно заметить задержку роста ребенка.
Синдром Клайнфельтера — наиболее часто встречающийся с типичной картиной синдром с набором 47, ХХУ. Включает случаи полисомии по половым хромосомам в кариотипе у мужчины при которых имеется не менее двух Х-хромосом и не менее одной У-хромосомы. Около 80% всех случаев заболевания связаны с присутствием в кариотипе дополнительной Х-хромосомы (47, ХХУ). У таких мальчиков можно обнаружить тельца Барра в 15-25% эпителиальных клеток слизистой щек, барабанные палочки». У них также определяются и Р-тельца в 65% лимфоцитов крови. У других пациентов с синдромом Клайнфельтера в основном регистрируются мозаичные формы, когда, например, часть клеток организма больного может содержать нормальный набор хромосом.
Больные с синдромом Клайнфельтера высокого роста. Часто встречаются диспропорционально длинные нижние конечности, сколиоз, деформация грудной клетки. У некоторых больных с этим синдромом выявлялись катаракты, снижение слуха, врожденные пороки сердца, варикозное расширение вен. Достаточно часто у мальчиков с синдромом Клайнфельтера регистрируются умственная отсталость легкой степени или неустойчивость внимания, повышенная утомляемость, повышенная внушаемость, снижение инициативности, незрелость суждений. Степень тяжести умственной отсталости нарастает с увеличением числа Х-хромосом в кариотипе больного. Так, больные с 49, ХХХХУ страдают глубокой олигофренией. И то же время интеллект у людей с 47, ХХУ может быть нормальным.
Синдром «кошачьего крика» — это частичная моносомия по короткому плечу хромосомы 5 (5р-). Заболевание было описано в 1963 г. Дж. Леженом. У детей с умственной отсталостью и необычным лицом было обнаружено уменьшение размера короткого плеча хромосомы из группы В. Причиной данного заболевания является делеция от 1/3 1/2 длины короткого плеча одной из 5-й пары хромосом (рис. 9.14). Соотношение мужчин и женщин составляет 1:1,3. Кариотип больного ребенка 46, XX, 5р- или 46, ХУ, 5р-. Синдром моносомии 5р- является самым частым среди заболеваний, вызванных структурным изменением хромосом. Частота синдрома достаточно высокая для структурных синдромов — на 1 новорожденного из 45 000.
У девочек данный синдром регистрируется несколько чаще. Клиническая картина синдрома моносомии 5р характеризуется необычным плачем, напоминающим требовательный кошачий крик или мяуканье, обусловленным сужением гортани, мягкостью хрящей, меньшими размерами надгортанника, необычной складчатостью слизистой оболочки. По этой причине этот синдром был назван синдромом «кошачьего крика».
Клиническая картина синдрома 5р у грудного ребенка характеризуется специфическим криком, напоминающим кошачий крик, обусловленный сужением гортани или патологией голосовых связок.
Почти у всех больных имеются различные изменения мозговой части лица и черепа. У больных отмечаются лунообразное лицо, гипертелоризм, микроцефалия, микрогения, эпикант, высокое нёбо, плоская спинка носа. Наружные уголки глаза располагаются ниже внутренних (антимонголоидный разрез). Мышечный тонус несколько снижен. С увеличением возраста ребенка специфический крик и мышечная гипотония постепенно исчезают. Кроме того, встречаются врожденные пороки сердца и некоторых других внутренних органов.
Вместе с тем становятся все более заметными задержка умственного и физического развития, микроцефалия (малые размеры черепа), обнаруживаются косоглазие, атрофия зрительных нервов, изменение сетчатки.
Приложение 3
Домашнее задание
1.Составление электронных презентаций по темам (на выбор):
1.«Проявление умственной отсталости при хромосомных синдромах»
2.«Половая функция при хромосомных синдромах»
3.«Группы риска по развитию хромосомных синдромов»
4. «Синдром трисомии 8»
5. «Синдром Вольфа-Хиршхорна»
6.«Синдром кошачьего глаза»
7. «Синдром Реторе»
8.«Синдром Орбели»
9.«Синдром де Груши»
10.Синдром трипло-Х
11.Синдром ХУУ
2.Тест
1. Классификация наследственных болезней человека в зависимости от вида мутаций, лежащих в их основе:
а) генные, хромосомные
б) соматические, индуцированные
в) индуцированные, генные
2. Методы диагностики генных наследственных болезней
а) биохимический, иммунологический
б) популяционно-статистический
в) дерматоглифики
г) моделирования
3. Тип наследования синдрома Марфана
а) аутосомно-рецессивный
б) аутосомно-доминантный
в) сцепленный с Х- хромосомой
г) голандрический
4. Какие мутации относятся к хромосомным:
а) делеция;
б) триплоидия;
в) инверсия;
г) изохромосома.
5.Какой кариотип характерен для больного с синдромом Эдвардса?
А) 46,XY,21
Б) 47,XXY
В) 47,XX,18
Г)46 ХО
6. Какие мутации относятся к геномным:
а) инверсии, транслокации, дупликации, делеции;
б) полиплоидии, анеуплоидии;
в) триплоидии, тетраплоидии;
г) внутрихромосомные и межхромосомные перестройки.
7.В каких возрастных интервалах существенно повышается риск рождения ребенка с хромосомными аномалиями?
А)20-25 лет
Б)25-30 лет
В)30-35 лет
Г)35-40 лет
8.Полиплоидия – это
А) уменьшение числа хромосом в наборе на несколько пар;
Б) диплоидный набор хромосом в гамете;
В) увеличение числа хромосом, кратное гаплоидному набору.
9.Каковы причины возникновения трисомий?
А) отставание хромосом в анафазе;
Б) нерасхождение хромосом;
В)Точечные мутации;
10. Укажите правильную формулу кариотипа при синдроме Шерешевского-Тернера:
а) 46,ХY,5р-;
б) 45,Х0;
в) 47,ХХХ, 47,ХХY;
г) 46,ХХ.
11. Укажите правильную формулу кариотипа при синдроме Патау:
а) 47,ХХ,18+;
б) 47,ХY,13+;
в) 46,ХХ,5р-;
г) 47,ХХY;
12. Низкий рост, крыловидная складка на задней поверхности шеи, слабо выражены вторичные половые признаки, бесплодны, интеллект не нарушен - симптомы заболевания
А)Шерешевского-Тернера
Б) синдром трисомии Х
В) комплекс УО
Г) синдром «кошачьего крика»
13. Заболевание, связанное с делецией короткого плеча 5 пары аутосом, называется
а) кретинизм
б) хорея Гентингтона
в) синдром Тей-Сакса
г) синдром «кошачьего крика»
14. Характерный плач ребенка, плоское лицо, пороки сердечно-сосудистой системы, скелета, почек, слабоумие – это симптомы
а) синдрома «кошачьего крика»
б) синдрома Дауна
в) синдрома Патау
г) синдрома Эдвардса
15. Низкая масса тела, избыточная кожа образует складки, деформации костей стопы, микроцефалия, микрофтальмия – это симптомы
а) синдром Эдвардса
б) синдрома Патау
в) синдрома Дауна
г) синдрома Тей-Сакса
16. Специфические фенотипические признаки синдрома Патау (выберете неверный вариант)
А)глубокая идиотия
Б)грыжа белой линии живота
В)Микроцефалия
Г)пороки развития головного мозга
17. Синдром Клайнфельтера это
А)Полисомия "Х" хромосомы в мужском кариотипе
Б)Моносомия "Х " хромосомы в мужском кариотипе
В)Моносомия "Х" хромосомы в женском кариотипе
18 Синдром Тернера это
А)Моносомия "ХО"
Б)Полисомия "ХХХ"
В)Полисомия "ХХУ"
Эталоны ответов:
1.А
2.А
3.Б
4.Б
5.В
6.Б
7.Г
8.В
9.Б
10.Б
11.Б
12.А
13.Г
14.А
15.А
16.Б
17.А
18.А
Список использованной литературы
-
Учебная литература: Гайнутдинов И.К., Юровская Э.Д. Медицинская генетика: Учебник. – М.: Издательско-торговая корпорация «Дашков и К, 2009. – 336 с.
-
Медицинская генетика: учебник / под ред. Н.П. Бочкова. – М.: ГЭОТАР-Медиа, 2019. – 224 с.: ил.














Вебинар для учителей

Свидетельство об участии БЕСПЛАТНО!
Полезное для учителя

Реализация образовательных программ осуществляется с применением исключительно электронного обучения и ДОТ



