
Россия, Чебоксары


СДЕЛАЙТЕ СВОИ УРОКИ ЕЩЁ ЭФФЕКТИВНЕЕ, А ЖИЗНЬ СВОБОДНЕЕ
Благодаря готовым учебным материалам для работы в классе и дистанционно
Скидки до 50 % на комплекты
только до 28.05.2025
Готовые ключевые этапы урока всегда будут у вас под рукой
Организационный момент
Проверка знаний
Объяснение материала
Закрепление изученного
Итоги урока
Была в сети 21.04.2025 00:03

Белова Виктория Геннадьевна
учитель биологии
42 года
Местоположение
Специализация
Проект по теме "Наследственные заболевания Чувашской Республики"
Категория:
Биология
05.06.2018 15:48

Просмотр содержимого документа
«Проект по теме "Наследственные заболевания Чувашской Республики"»
Муниципальное бюджетное учреждение
«Средняя общеобразовательная школа №53 с углубленным изучением
отдельных предметов» города Чебоксары Чувашской Республики
Проект по теме:
«НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ ЧУВАШСКОЙ РЕСПУБЛИКИ»
Проект выполнили:
обучающиеся 10А класса
Научный руководитель:
Белова Виктория Геннадьевна,
учитель биологии
Чебоксары, 2018
Содержание
Введение …………………………………………………………………………………3
Цель и задачи проекта ………………………………………………………………......4
Актуальность проекта ……………………………………………………………….......4
Теоретическая часть проекта
1.1. Понятие наследственные и врожденные заболевания ………………….…….......4
1.2. Медико-генетическое консультирование в Чувашии ………………………......4-5
1.3. Причины возникновения наследственных заболеваний …………………….....6-7
1.4. История изучения наследственных заболеваний ………………………………7-8
1.5. Статистика по наследственным заболеваниям в Чувашии ………………..........8-9
1.6. Описание наследственных заболеваний ……………………………………… 9-21
Практическая часть проекта
Решение генетических задач на наследственные заболевания ………………….21-30
Заключение …………………………………………………………………………30-31
Источники использованной литературы …………………………………………….31
Интернет ресурсы ……………………………………………………………………..31
Введение
В последние годы в связи с бурным развитием медицинской генетики меняются традиционно сложившиеся в прошлом веке представления об этиологии и патогенезе многих заболеваний. Значительно возрос вклад наследственной патологии в структуру заболеваемости и смертности, как взрослого, так и детского населения. При относительной редкости отдельных форм наследственных болезней их общая частота в популяции человека достаточно высока. В связи с этим все большее значение приобретают знания и мероприятия, направленные на изучение частоты встречаемости, механизмов распространения и профилактики наследственной патологии.
По данным Всемирной Организации Здравоохранения 5-7% новорожденных имеют различную наследственную патологию. Число зарегистрированных наследственных болезней постоянно растет. К настоящему времени согласно международной классификации выделено 17905 описаний различных фенотипов и болезней, из которых 16792 с аутосомным типом наследования (аутосомно-доминантный и аутосомно-рецессивный), 994 – с X-сцепленным, 56 – с Y-сцепленным и 63 – с митохондриальным. Из такого разнообразия описаний примерно 5000 - 6000 относятся к наследственным болезням и синдромам.
Цель проекта:
рассмотреть различные наследственные заболевания человека, в том числе характерные и для Чувашии и выявить причины их появления
Задачи проекта:
Предметные: сформулировать понятия о наследственных заболеваниях Чувашии, их характерных особенностей, причин возникновения, мер профилактики и возможности лечения
Метапредметные: развить умение работать в группе с раздаточным материалом, выявление по тексту необходимой информации, умение анализировать полученную информацию и выступать перед аудиторией, умение составлять и решать генетические задачи по данной тематике
Личностные: сформировать у учащихся толерантное отношение к людям с наследственными заболеваниями, повысить интерес к изучаемому предмету.
Актуальность работы:
Актуальность настоящей работы заключается в том, что медико-генетические исследования по изучению наследственных заболеваний в Чувашской Республике проводятся редко. Вместе с тем чуваши по численности занимают в России пятое место, а некоторые события в истории чувашей, с точки зрения популяционной генетики, способствовали накоплению в популяции редких генов, в том числе генов наследственных болезней, которые мы и изучили в нашем проекте.
Теоретическая часть проекта
Понятие «наследственные» и «врожденные» заболевания
Во всем мире ученые бьют тревогу по поводу стремительно ухудшающейся экологической обстановки, последствия которой мы все больше и больше ощущаем на себе. Особенно чувствительна к многочисленным вредностям будущие матери, внутри которых уже зародилась и развивается новая жизнь. К сожалению, неблагоприятные экологические воздействия на организм беременной женщины все чаще вызывают появление детей с врожденными пороками развития. Сейчас мы докажем, что врожденные и генетические (наследственные) болезни — совершенно разные понятия.
При врожденном заболевании человека в отличие от наследственного исходные яйцеклетка, сперматозоид и образовавшийся в результате их слияния зародыш были здоровы, но в результате негативного воздействия фактора внешней среды произошло нарушение формирования и развития какого-либо органа или системы.
Наследственные заболевания новорожденных имеют принципиально иное происхождение. Они связаны с поломкой какого-либо гена в половой клетке одного или обоих родителей. Яйцеклетка содержит 23 хромосомы, столько же хромосом находится внутри сперматозоида. При слиянии половых клеток отца и матери образуется новая клетка, содержащая полную информацию о будущем ребенке, а именно несколько десятков тысяч генов, каждый из которых отвечает за тот или иной признак: цвет глаз, волос, форму ушей, группу крови, резус-фактор, рост и т. д.
Наследственная болезнь возникнет, если встретились два одноименных патологических гена от матери и от отца. При этом родители являются совершенно здоровыми. Наследственные болезни не поддаются лечению, поэтому главное значение придается их предупреждению до наступления беременности или на ранних ее стадиях. Если супруги знают о наличии в генеалогическом древе родственников, страдающих хромосомными болезнями (гемофилия, фенилкетонурия, сахарный диабет, психические болезни, миопатия Дюшенна, хорея Хантингтона), они обязательно должны попасть на консультацию к генетику, главная задача которого — предупредить появление на свет больного ребенка.
Медицинское обследование будущих родителей начинается с изучения родословной, наличия наследственных заболеваний и состояния здоровья обоих супругов. Затем производится анализ хромосомного набора соматической клетки, по которому определяется наличие дефектных генов. Такие гены (до 10-12) есть у каждого, но ребенок родится больным в том случае, если встретятся два одноименных патологических гена — от отца и от матери. Если первый ребенок в семье страдает наследственным заболеванием, перед повторной беременностью обязательно следует пройти медико-генетическое обследование.
1.2. Медико-генетическое консультирование в Чувашии
Медико-генетическая консультация существуетв России с 1990 года, вначале был открыт консультативный кабинет медицинского генетика, позже были организованы биохимическая и цитогенетическая лаборатории. В цитогенетической лаборатории проводилось исследование кариотипа, в биохимической лаборатории проводились биохимический скрининг мочи (уринолизис), неонатальный скрининг на фенилкетонурию.
С 1994 года неонатальный скрининг начали проводить в Межрегиональном Медико - генетическом центре г. Нижний Новгород.
С 2000 года медико-генетическая консультация вошла в состав Центра планирования семьи и репродукции, а с октября 2001 года в состав ГУЗ «Президентский перинатальный центр» в Чувашской Республики.
Медико-генетическая консультация состоит из клинического и лабораторного отделов.
Консультативный прием ведут 2 врача-генетика: генетик-педиатр, который занимается семьями, в которых дети с подозрением на наследственную патологию; генетик-акушер-гинеколог, ведет в основном прием беременных, проводит подготовку к проведению пренатальной диагностики и участвует в проведении пренатальной диагностики.
Медико-генетическая консультация тесно сотрудничает с Федеральными центрами г. Москва, г. С.-Петербург, проводятся при необходимости консультации со специалистами, заочные и по телемедицине, для уточнения диагноза наследственной патологии проводится ДНК-диагностика в лаборатории ДНК-диагностики г. Москва.
Организована работа совместно с кабинетами планирования семьи, кабинетом по невынашиванию беременности проведение профилактики врожденных пороков развития и наследственных заболеваний, разработан план обследования супружеской пары и проведения лечения перед наступлением беременности. Проводится расчет риска рождения ребенка с наследственной патологией индивидуально для каждой пары.
Основные задачи Медико-генетической консультации:
- уточнение диагноза наследственной патологии
- профилактика наследственной патологии наследственных заболеваний и врожденных пороков развития, проведение профилактики врожденных пороков развития и наследственных заболеваний.
Показания к направлению в медико-генетическую консультацию:
-уточнение диагноза наследственного заболевания после рождении ребенка ( подозрение на синдром Дауна)
-рождение ребенка с врожденными пороками развития.
-отставание ребенка в физическом и половом развитии
-нарушение половой дифференцировки, первичная аменорея в сочетании с недоразвитием вторичных половых признаков
-наличие у ребенка задержки нервно-психического развития
-хронические прогрессирующие заболевания неясного генеза и неподдающиеся обычной терапии
-непереносимость лекарственных препаратов, пищевых продуктов
-нарушения поведения ребенка, неадекватность, агрессивность, аутизм
-наличие у ребенка судорожного синдрома, не связанного с натальной травмой, неподдающегося противосудорожной терапии
-длительная желтуха у ребенка неясного генеза
-отягощенный акушерский анамнез (мертворождение, рождение ребенка с ВПР, с наследственной патологией в анамнезе, преждевременные роды, невынашивание беременности, бесплодный брак).
-кровнородственный брак
-беременные, у которых отягощенный акушерский анамнез
-беременные, старше 35 лет
-беременные, работающие на вредном производстве, принимавшие на ранних сроках беременности лекарственные препараты, проводили рентгенологические обследования, переболели вирусной инфекцией в первом триместре беременности.
Причины возникновения наследственных заболеваний
Врожденная патология, в первую очередь врожденные пороки развития являются серьезной медицинской и социальной проблемой во всем мире.
В настоящее время по статистике Всемирной организацией Здравоохранения риск рождения ребенка с врожденными пороками развития и наследственной патологией составляет 5-6 % для супружеской пары до 35 лет. Больше половины пороков развития обусловлены действием факторов внешней среды или сочетанным действием наследственных и средовых факторов. Основные причины возникновения пороков развития по данным ВОЗ:
- пожилой возраст женщины
- самолечение и бесконтрольный прием лекарственных препаратов во время беременности
- наличие патологии (сахарный диабет, гипертоническая болезнь, заболевания щитовидной железы и др.)
- переболевание во время беременности вирусными заболеваниями (краснуха, грипп, ОРВИ и др.).
-проведение во время беременности вакцинации от кори, краснухи и др.
- профессиональные вредности
- недостаточное, несбалансированное питание
- недостаток фолиевой кислоты
- стрессовые ситуации
- алкоголь
- курение
- недостаточный перинатальный контроль.
Периконцепционная профилактика заключается в обеспечении оптимальных условий для созревания половых клеток, раннего развития плода и включает в себя систему мероприятий, направленных на устранения некоторых факторов риска и широком смысле улучшение здоровья будущих родителей.
История изучения наследственных заболеваний

Наследственность как свойство всех организмов интересовала людей с древних времен. Но только в XIX в это явление стали объективно изучать. Действительно научный вклад внес Грегор Мендель – основатель научной генетики. В 1866 г он опубликовал результаты экспериментов, в которых показал, что наследственность передается через половые клетки от одного поколения к другому, не смешиваясь, не растворяясь. Его опыты заложили основу концепции гена, которая сохраняет свое ведущее место до настоящего времени.
Законы Менделя заново переоткрыли и приняли в 1900 г Хьюго де Фриз из Голландии, Корренс из Германии, Чермак из Англии. Этот год считается годом рождения генетики как науки.
В России в середине XIX в над проблемами генетики работал Флоринский В.М., который указывал на значение среды в формировании наследственных признаков, вред близкородственных браков, наследственный характер пороков развития, глухонемоты, альбинизма.. В конце XIX в англичанин Ф.Гальтон англичанин подтверждал эти понятия.
В 1900г Ландштейнер описал систему групп крови АВО, наследование которых подтвердилось работами Гиршфельда и Дунгерна.
В 1924 г Берштейн установил, что система АВО контролируется серией множественных аллелей одного локуса.
В первом десятилетии XX в было обнаружено наследование нарушений обмена веществ. В 1902 г английский врач Гэррод изучая наследственные болезни, сделал вывод, что нарушение обмена веществ наследуется по законам Менделя – им создано направление генетики – биохимическая генетика человека. Исследуя одну из наследственных болезней обмена веществ — алкаптонурию, он предположил, что в ее основе лежит генетически обусловленное врожденное нарушение обмена веществ, и что таким же образом могут развиваться и другие наследственные болезни. Дальнейшее понимание природы наследственных болезней связано с успехами в изучении механизма реализации генетической информации. Вехой в этом направлении явилась сформулированная Дж. Бидлом и Тейтом концепция «один ген — один фермент», означавшая, что гены контролируют синтез ферментов, и объяснявшая механизм возникновения наследственных врожденных нарушений обмена веществ, описанных Гэрродом.
1919 г организована кафедра генетики Ю.А.Филипченко в Петроградском университете, а в то же время Вавилов Н.И. сформулировал закон гомологических рядов в наследственной изменчивости.
В первые десятилетия XX века роль наследственности в формировании поведения человека и наследственной отягощенности населения была даже существенно преувеличена.
Огромная роль в развитии медицинского направления в генетике принадлежит основоположнику клинической генетики С. Н. Давиденкову одновременно генетику и невропатологу. Наряду с огромным вкладом в изучение генетики нервных болезней он на несколько десятилетий определил разработку общегенетических проблем. Он первым в мире поставил вопрос о необходимости составления каталога генов человека, сформулировал понятие о генетической гетерогенности наследственных болезней, организовал медико-генетическую консультацию. С 1930-1937 гг. медицинская генетика развивалась в Медико-биологическом институте. Однако в период сталинских репрессий генетика была объявлена "лженаукой".
В конце 1940-х гг. Л. Полинг (совместно с сотрудниками) обнаружил аномальное поведение при электрофорезе гемоглобина, полученного от больных серповидноклеточной анемией.
В 1944 г было установлено, что передача наследственной информации связана с ДНК
Генетические исследования у нас в стране возобновились только в начале 60-х годов. При этом в мире, начиная с 50-х годов XX в, наступает наиболее бурный период развития генетики человека.
С 1956 г начало научного направления - цитогенетики, т.е. изучение хромосом, Леван и Тио, а также Форд и Хамертон независимо друг от друга установили, что количество хромосом у человека 46.
В 1957 г В. Ингрем доказал, что этот гемоглобин является мутантным, а его аномальные свойства обусловлены специфическим замещением в его молекуле одного из аминокислотных остатков (глутаминовой кислоты) на другой (валин). В результате было сформулировано представление о молекулярных болезнях, в основе которых лежат изменения последовательности нуклеотидов (мутации) в гене и соответствующие им изменения аминокислотной последовательности кодируемого этим геном белка.
В 1959 г. была открыта хромосомная природа болезней, и цитогенетика на несколько лет стала ведущим направлением. В этот период сформировалась клиническая генетика как результат слияния цитогенетики, менделевской генетики и биохимической генетики, а человек стал главным объектом общегенетических исследований. К настоящему времени обнаружено около 1000 вариантов патологических изменений хромосом у человека и уже возможна ДНК диагностика более 100 наследственных патологий.
1.5. Статистика по наследственным заболеваниям в Чувашии
Разнообразие аутосомно-доминантных заболеваний у населения Чувашской Республики
С распространенностью 1:50000 и чаще в Чувашии зарегистрировано 15 заболеваний: вульгарный ихтиоз (1:3148), ладонно-подошвенная кератодермия (1:13917), нейрофиброматоз (1:11497), несиндромальная нейросенсорная тугоухость (1:14690), наследственная моторно-сенсорная нейропатия (1:17628), брахидактилия тип В (1:22035), гипохондроплазия (1:23780), постаксиальная полидактилия (1:33052), доминантная олигофрения (1:33052), эктрадактилия (1: 37774), врожденная катаракта (1:44070), экзостозная хондродисплазия (1:44070), синдромы Элерса-Данло (1:37774), синдром Марфана (1:26442) и арахнодактилия с деформацией пальцев (1:37774).
Разнообразие аутосомно-рецессивных заболеваний у населения Чувашской Республики
К частым аутосомно-рецессивным заболеваниям (с распространенностью 1:50000 и чаще) можно отнести 10 болезней: нейросенсорную тугоухость (1:4334), пигментный ретинит (1:11498), врожденный гипотрихоз (1:13221), недифференцированную олигофрению (1:14690), врожденную катаракту (1:17628), микроцефалию с олигофренией (1:22035), микрофтальм с микрокорнеа (1:44070), врожденный гипотиреоз (1:23780), гипофизарный нанизм (1:24319) и ихтиозиформную эритродермию (1:37774).
Разнообразие Х-сцепленных заболеваний у населения Чувашской Республики
Спектр заболеваний, сцепленных с Х-хромосомой представлен 11 формами. Частыми, т.е. с распространенностью 1:50000 мужчин и чаще, выявлено 6 заболеваний: олигофрения (1:6610), гемофилия А (1:22035), ихтиоз (1:26442), наследственная моторно-сенсорная нейропатия, нистагм и хореодеремия с распространенностью 1:44070.
1.6. Описание наследственных заболеваний
Наследственные заболевания - это заболевания, возникновение и развитие которых связано с различными дефектами и мутациями. Нередко причиной возникновения врождённых заболеваний становиться ареал проживания, давайте рассмотрим некоторые заболевания, которые могут проявляться в Чувашской Республике.
В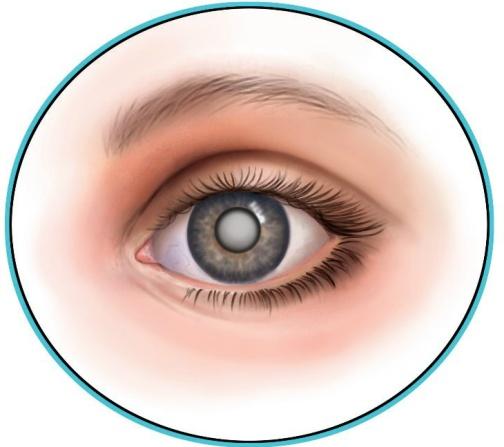 рождённая катаракта
рождённая катаракта
Катаракта – это его помутнение, следствием которого является снижение зрения. Обычно это заболевание является возрастным, но может встречается также и с рождения. В Чувашии такая вероятность составляет 1 к 16 тысячам.
Название этой болезни дал Гиппократ. В греческом языке катарактес – это водопад. Название такое потому, что с этой болезнью человек видит как сквозь толщу воды. А в 16 веке Галлена Паулус Агина определил расположение катаракты и описал ее отличия от глаукомы. Катаракту он представлял как водяное образование, а ухудшение зрения связывал с нарушениями в работе зрительного нерва. Ученый определил появление катаракты преимущественно у пожилых людей, перенесших травму или страдающих другими глазными заболеваниями.
Катаракты врожденные могут передаваться по наследству и, судя по статистике, в 70% случаев передается по мужской линии. Но также могут быть вызваны патологией беременности: поздний возраст матери; резус-несовместимость матери и плода; перенесённая болезнь матери на ранних сроках (более опасна краснуха)
Врожденная катаракта на ранней стадии не даёт о себе знать ярко выраженными симптомами, и поначалу больной ребенок внешне может ничем ни отличаться от здоровых детей.
Впоследствии появляются первые признаки: различные затемнения в районе зрачков, неспособность закреплять взгляд на конкретных предметах и лицах людей, косоглазие, постоянное использование одного и того же глаза при рассматривании предметов или игрушек, подергивание глазных яблок.
Внешние признаки могут быть неубедительными, поэтому в целях подтверждения или опровержения диагноза врач-офтальмолог должен провести ряд исследований, которые точно помогут выявить все имеющиеся отклонения, связанные со зрительными органами. К таким исследованиям относят: офтальмоскопию, проверка глазного яблока при помощи ультразвука, щелевая биомикроскопия, исследование глазных тканей.
Лечение врождённой катаракты
Рассматриваемая болезнь хрусталика не во всех случаях влечёт за собой резкое ухудшение и, тем более полную потерю зрения. Нет необходимости проводить операцию по удалению больного участка глаза, если центральная зрительная функция в норме, и нет факторов, препятствующих дальнейшему развитию зрительной системы. Вместо этого, чтобы быть и оставаться зрячим человеком, необходимо соблюдать специальный курс медикаментозной терапии, который остановит прогрессирование заболевания.
Если же катаракта имеет сильные признаки, то проводится операция. Почти во всех лечебных учреждениях для лечения катаракты проводят операцию под названием факоэмульсификация. Принцип факоэмульсификации состоит в разрушении естественного помутневшего хрусталика с помощью ультразвука. Для этого в глаз через крохотный разрез в роговице вводится специальный зонд, который испускает ультразвуковые волны. После разрушения хрусталика его частицы вымываются из глаза. На его место вводится искусственная интраокулярная линза (ИОЛ). Длительность такой операции составляет, как правило, 15-20 минут, она легко переносится пациентами.
Г ипофизарный нанизм
ипофизарный нанизм
Гипофизарный нанизм (карликовость) – это заболевание, которое характеризуется резким отставанием в росте и физическом развитии, связанном с абсолютной или относительной недостаточностью гормона роста. Карликовым принято считать рост у мужчин от 130 см, а у женщин – от 120 см. В то время как социально-приемлемый рост для девочек он составляет 155 см, а для мальчиков – 165 см. В Чувашии вероятность родиться с таким заболеванием составляет 1 к 24 тысячам .
Врожденный недостаток гормона роста – возникает из-за наследственности; аномалий в строении и развитии гипоталамуса или гипофиза; недостаточной выработке в гипоталамусе стимуляторов синтеза соматотропного гормона( гормона роста). При рождении у ребенка отсутствуют отклонения в параметрах физического развития. Только в 2-3 года дети начинают резко отставать в росте от своих сверстников, при этом все части тела ребенка остаются пропорционально развитыми – они только перестают расти.
Возможно ли вылечить нанизм?
Если нанизм связан с дефицитом гормона роста, то лечение нужно целесообразно проводить в тот момент, пока не закрылись костные зоны роста, то есть до момента полового созревания. Детям прописывается гормон роста. Обязательным условием лечения является правильное питание и прием необходимых витаминов. За первый год лечения гормоном роста ребенок может подрасти сразу на 10-12 см, причем максимальный рост обозначится в первые полгода терапии. Потом темпы роста снижаются, но дети все равно растут лучше, чем без лечения. Чем раньше начинается терапия, тем больше шансов у ребенка догнать своих сверстников по показателям физического развития.
Гипотрихоз
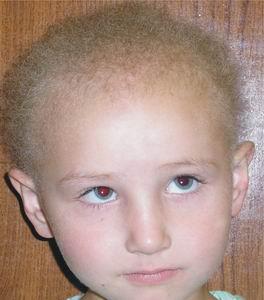 Гипотрихоз - это достаточно редкое заболевание, проявление его заключается в полном либо частичном выпадении волос на всём теле или на отдельных участках. Мы расскажем вам про врождённый гипотрихоз, который является генетической аномалией, передающейся по наследству. Заболевание, как правило, поражает всех членов семьи, связанных кровным родством. Согласно статистике в Чувашии из 1300 рождённых детей 1 ребенок болеет гипотрихозом.
Гипотрихоз - это достаточно редкое заболевание, проявление его заключается в полном либо частичном выпадении волос на всём теле или на отдельных участках. Мы расскажем вам про врождённый гипотрихоз, который является генетической аномалией, передающейся по наследству. Заболевание, как правило, поражает всех членов семьи, связанных кровным родством. Согласно статистике в Чувашии из 1300 рождённых детей 1 ребенок болеет гипотрихозом.
Симптомы у детей в большинстве случаев проявляются вскоре после рождения. Полное отсутствие волос — явление редкое. Чаще всего волосяной покров сформирован, но при этом он достаточно редкий, имеет залысины, а растущие волосы очень тонкие и ломкие. Излечить этот недуг практически невозможно. Облысение — это внешнее проявление гипотрихоза. Причиной же его является либо аномальное строение кожи, либо генетические отклонения в работе эндокринной системы. Это приводит к тому, что волосяные фолликулы полностью или частично атрофированы, либо функционируют неполноценно, образуя ломкие и тонкие волосы.
В большинстве случаев врождённый гипотрихоз у больного является лишь симптомом серьезных генетических отклонений. Наряду с ним, человек часто страдает от хронического ринита, у него неправильно функционируют сальные железы, недоразвиты ногтевые пластины, встречаются также и отставания в умственном развитии. Встречаются также случаи отрастания жестких, толстых и коротких волос.
Причины возникновения и способы лечения гипотрихоза:
Гипотрихоз является достаточно редким заболеванием, которое передается по аутосомно-доминантному типу. Излечить врождённый гипотрихоз невозможно. Рекомендуется избегать травматизации волос и желательно употреблять больше препаратов цинка, фитина, витаминов, гормональных препаратов
Альбинизм

Альбинизм — это наследственное заболевание, связанное с нарушением пигментного обмена в организме. В Чувашии встречаемость альбинизма среди детей составила 1:16965.
При этой патологии возникает дефицит меланина — особого вещества, придающего окраску коже, пигментной и радужной оболочкам глаза, волосам, ногтям. Казалось бы, что такого, если дело всего лишь в цвете кожи, глаз, волос? Но не все так просто на самом деле. Отсутствие или снижение содержания меланина приводит к возникновению не только косметических дефектов, но и ряда проблем со здоровьем. В частности, это непереносимость солнечных лучей, нарушение зрения. У альбиносов повышен риск развития рака кожи. Альбинизм не является болезнью, угрожающей жизни пациента, однако может существенно влиять на ее качество. Из этой статьи Вы сможете узнать подробнее о том, что такое альбинизм, почему он возникает, как проявляется у человека и чем лечится.
Причины возникновения альбинизма:
У человека цвет кожи, волос, бровей, ресниц, радужной оболочки глаза определяется наследственностью. Это означает, что каждый из нас рождается с запрограммированным цветом кожи и ее придатков, цветом глаз. Истинная окраска кожи определяется на участках, которые не подвергаются воздействию солнечных лучей (область ягодиц). Альбинизм — это результат генных нарушений, причем самых разнообразных. В организме человека существует несколько генов, определяющих цвет выше перечисленных структур. Любая мутация хотя бы в одном из них приводит к нарушению содержания пигмента меланина и, соответственно, к возникновению альбинизма. Это означает, что альбинизм — болезнь врожденная, хотя и не всегда сразу проявляющаяся. Альбинизм может передаваться по наследству последующим поколениям, причем как доминантно, так и рецессивно (то есть проявлять себя во всех поколениях или только при совпадении двух патологических генов). Сама по себе мутация может возникнуть спонтанно, то есть при отсутствии подобной проблемы в роду.
Меланин (от слова «melanos», что в переводе означает «черный») — это вещество, окрашивающее кожные покровы в определенный цвет. Чем больше меланина, тем темнее окраска. Степень дефицита меланина определяет выраженность альбинизма. При тотальном отсутствии пигмента альбинизм является полным. Образование меланина зависит от фермента тирозиназы (именно ее содержание и активность определяются генетически). Под воздействием тирозиназы из тирозина в клетках кожи и радужной оболочки глаза образуется меланин. Меланин защищает человека от ультрафиолетового облучения путем формирования загара. Меланин в глазу обеспечивает проникновение света только через зрачок, что позволяет нам безболезненно смотреть на яркий свет, а также защищает глаз от видимой и длинноволновой части лучистой энергии.
В медицине принято различать две большие клинические группы этой патологии:
- полный альбинизм, или глазо-кожный;
- частичный альбинизм, или глазной.
Полный альбинизм развивается в тех случаях, когда генетический дефект определяет содержание меланина и в коже, и в волосах, и в зрительной системе.
Частичный альбинизм означает нехватку меланина только в зрительной системе.
Полный, или глазо-кожный альбинизм:
Люди, страдающие этим видом альбинизма, с рождения имеют белую кожу , белые волосы и голубые радужки. Это связано с полным отсутствием тирозиназы. Поскольку радужка полностью пропускает свет, то сам глаз приобретает красноватый или розовый оттенок (из-за просвечивающихся сосудов), что формирует феномен «красных глаз». Из-за полного отсутствия пигмента, кожа таких больных не способна бороться с ультрафиолетовым излучением, загар не формируется. Под воздействием солнечных лучей люди с глазо-кожным альбинизмом сразу получают ожог. Именно поэтому они вынуждены прятать свои кожные покровы с помощью одежды с длинными рукавами, постоянно носить брюки или длинные юбки, шляпы. У этих альбиносов существенно выше риск развития рака кожи. Кожа на протяжении всей жизни остается белой, никакие пигментации не образуются. Имеется склонность к возникновению сосудистых звездочек и кератом. Обычно кожные покровы сухие, с низкой продукцией пота. Волосы у альбиносов тонкие и мягкие.
Глазные проявления заключаются не только в красноватом оттенке глаз. Таким людям трудно переносить яркий свет, поэтому для них характерна светобоязнь. Острота зрения обычно снижена, часто наблюдается нистагм — колебательные непроизвольные движения глазных яблок. Существует разновидность глазо-кожного альбинизма, при которой тирозиназа отсутствует не полностью, а лишь частично (или ее активность снижена). В этом случае развивается неполный альбинизм (альбиноидизм). Клинически это проявляется изменением цвета кожи от белого (при значительном снижении активности тирозиназы) до практически нормального (при почти нормальной активности фермента). При рождении у таких людей кожа белая, глаза с красноватым оттенком, волосы без пигмента. Однако по мере взросления у людей с этой разновидностью альбинизма кожа темнеет (и может немного загорать), волосы накапливают желтый пигмент. Радужная оболочка также пигментируется: появляется светло- или темно-коричневая пигментация, иногда участками, а не тотально. У взрослого человека с такой разновидностью альбинизма цвет глаз может быть любым, даже темно-карим. Так что, далеко не все альбиносы имеют красные глаза, как принято считать. Могут появляться веснушки и пигментные пятна. При этой разновидности альбинизма брови и ресницы имеют более темный цвет, чем волосы на голове. Острота зрения снижена, однако может увеличиваться с возрастом (по мере потемнения радужки). Еще одна довольно распространенная разновидность альбинизма среди африканского населения — это рыжий глазо-кожный альбинизм. В данном случае генетический дефект обусловливает производство меланина не черного цвета, а коричневого. У таких больных кожа рыжеватого оттенка или светло-коричневая, а радужки коричнево-голубого цвета.
Частичный, или глазной альбинизм:
Эта форма альбинизма имеет клинические проявления только со стороны органа зрения. Кожа пигментирована нормально, способна загорать. Волосы обычные и по цвету, и по структуре.
Эритроцитоз
Эритроцитоз — это патологическое состояние, при котором количество эритроцитов и гемоглобина в крови увеличивается. Кровь при этом становиться вязкой, что затрудняет её движение по сосудам и нарушает кислородный обмен. При отсутствии лечения в организме могут возникнуть необратимые изменения.
Такой синдром не является отдельным заболеванием, и возникает вследствие адаптации организма к различным аномальным процессам. В некоторых случаях эритроцитоз свидетельствует о болезни, спровоцировавшей избыточное производство эритроцитов кроветворными органами. Поэтому прежде чем лечить сам симптом, следует выяснить причины его возникновения. 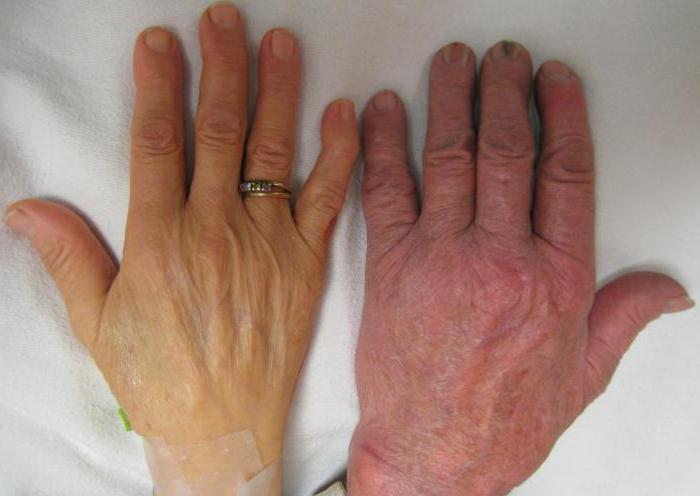
Формы эритроцитоза
В медицинской практике выделяют три формы такого патологического состояния. И у каждой из них есть свои причины развития.
Первая форма – это истинный эритроцитоз. Встречается он достаточно редко и причины его развития кроются в дефектных генах и наследственности. При такой форме происходит разрастание костного мозга, который продуцирует большое количество эритроцитов, что приводит к увеличению их концентрации в крови. Есть и другие причины развития истиной формы патологического состояния. В частности, он может возникнуть у людей, отравившихся угарным газом или же у тех, кто имеет различные патологии дыхательных путей, связанные с гипоксией, то есть недостаточным поступлением кислорода в кровь.
Вторая форма нарушения – абсолютный эритроцитоз, который также называют вторичным. При этой форме разрастание костного мозга не происходит, а увеличение количества красных кровяных телец связываются с различными патологическими процессами в организме человека. Причины, вызвавшие вторичный эритроцитоз, могут быть следующими: онкологические заболевания; доброкачественные опухолевидные процессы; длительная гипоксия (например, у курильщиков); болезни лёгких и лёгочная недостаточность; гемоглобинопатия. Причины, вызвавшие эту форму нарушения, бывают не только патологическими, но и физиологическими. Так, если человек проживает в высокогорной местности, где в недостаточном количестве получает кислород, его кровь становится более густой, а количество эритроцитов на литр возрастает. Кроме того, вызвать такое состояние, как эритроцитоз, может работа с определёнными веществами, способными связывать гемоглобин. Например, таким веществом является анилин.
Третья форма, причины которой связаны с уменьшением содержания воды в крови – это относительный эритроцитоз. Такое состояние может возникнуть внезапно и носить характер острого или хронического нарушения. Причины, вызывающие его, это: обезвоживание, вызванное различными причинами, в том числе длительной диареей или рвотой; большая потеря крови при открытых или внутренних кровотечениях; отравление токсическими веществами; гипертонический криз. Также данная форма патологического состояния встречается у людей, страдающих ожирением в той или иной степени, или же у тех, кто употребляет диуретики, увеличивающие мочевыделение. Кроме того, относительная форма может возникнуть как временное явление при стрессовых ситуациях и психических нарушениях. В таком случае после прекращения воздействия стрессового фактора уровень эритроцитов в крови возвращается в норму.
Первое появления эритроцитоза
Впервые эритроцитоз описан у членов одной семьи в 1908. В последующем были обнаружены у детей и взрослых в отдельных семьях у лиц разной национальности и в разных странах. Заболевание выявляется преимущественно в детском и юношеском возрасте.
Выделяют эритроцитоз с аутосомно-доминантным типом передачи и аутосомно-рецессивным типом передачи. Наиболее часто регистрируется семейный наследственный эритроцитоз с аутосомно-доминантным типом передачи. Причины, вызывающие эритроцитоз при данной патологии, обусловлены наличием аномального гемоглобина с увеличенным сродством к кислороду. Это происходит в результате замены некоторых аминокислот в альфа и бета цепях гемоглобина.
Другим вариантом эритроцитоза с аутосомно-доминантным типом передачи является эритроцитоз, развивающийся в результате нарушенного метаболизма.
Другая форма семейного наследственного эритроцитоза — это с аутосомно-рецессивным типом передачи. Это заболевание, характеризующееся эритроцитозом, нормальным количеством лейкоцитов и тромбоцитов, увеличенной продукцией эритропоэтина, но она встречается редко. Заболевание выявляется преимущественно в детском и юношеском возрасте. Случаи врожденной полицитемии с аутосомно-рецессивным типом наследования редки и встречаются , как правило спорадически. В мировой литературе описано небольшое число случаев этого заболевания. Выявлена данная патология у чуваш, марийцев, мордовин. У лиц других национальностей такой формы не выявлено. У лиц чувашской национальности семейный наследственный эритроцитоз имеет свои особенности. Описана данная форма и среди детей США, генетические корни которых имеют отношение к чувашской национальности.
Клиника этих случаев не совпадала ни с одной из известных нозологических форм, для которых были известны мутации в генах, приводящие к заболеванию. В связи с этим были предприняты клинические и генетические исследования.
Мутация, приводящая к развитию эритроцитоза у чувашей, выявлена в гене VHL, который является геном-супрессором опухолевого роста. При мутациях в нем у больных с синдромом VHL развиваются опухоли. Однако, ни у больных эритроцитозом (гомозиготных носителей мутации), ни у их родственников гетерозиготных носителей опухоли не обнаружены. Возможно, у них присутствует изменение в другом гене, являющимся модификатором, которое не позволяет развиться опухолям.
Остеопетроз
М раморная болезнь или остеопетроз (другие названия гиперостотическая дисплазия, болезнь Альберс-Шенберга, врожденный остеосклероз) – редкая врожденная патология опорно-двигательного аппарата, характеризующаяся избыточным формированием костной ткани.
раморная болезнь или остеопетроз (другие названия гиперостотическая дисплазия, болезнь Альберс-Шенберга, врожденный остеосклероз) – редкая врожденная патология опорно-двигательного аппарата, характеризующаяся избыточным формированием костной ткани.
Причины патологии
При врожденном остеосклерозе отмечается уплотнение и одновременная ломкость костей, функциональная недостаточность функций кроветворения, протекающих в костном мозге.
В организме здорового человека процессы формирования костной ткани остеобластами (клетки, продуцирующие костную ткань) и ее резорбции остеокластами (клетки, разрушающие костную ткань) находятся в равновесном состоянии.
Мраморная болезнь сопровождается нарушением функции остеокластов. При этом количество клеток-остеокластов может быть нормальным, повышенным либо пониженным. Точный механизм развития патологии до сих пор до конца не изучен. Доказано лишь то, что в развитии заболевания принимают участие 3 гена, ответственные за формирование костной и кроветворной ткани, существует наследственная предрасположенность к патологии: в большинстве случаев гиперостотическая дисплазия встречается у родственников.
Результатом генных нарушений при остеопетрозе становится снижение резорбции костной ткани, обусловленное недостатком фермента карбоангидразы в остеокластах. При этом интенсивность процессов формирования костей остеобластами не снижается, что приводит к уплотнению костной ткани, ее избытку.
Характерные проявления болезни
Постепенное вытеснение плотными костями костного мозга (вещества, содержащегося внутри кости и принимающего участие в Мраморная болезньпроцессах кроветворения) приводит к развитию тромбоцитопении, анемии, кроветворным процессам вне костного мозга – в печени,
лимфатических узлах, селезенке, в результате чего происходит их увеличение.
Основными симптомы мраморной болезни являются повышенная плотность и одновременная ломкость костей, склонность к переломам (особенно часто отмечаются переломы бедренной кости).
Самыми распространенными признаками болезни являются:
• увеличение печени, селезенки, лимфатических узлов;
• гипохромная анемия;
• гидроцефалия;
• нарушение зрительной функции, вплоть до слепоты (вызвано сдавлением
зрительного нерва).
Признаки развития
По времени развития патологии остеопетроз классифицируется на две формы:
• Ранняя форма мраморной болезни развивается у детей, характеризуется злокачественным течением и чаще заканчивается летальным исходом.
• Поздняя форма отмечается в зрелом возрасте, протекает легче, обычно бессимптомно, диагностируется по результатам рентгенологических исследований.
При ранней форме остеопетроза отмечаются боли в ногах, при ходьбе и физических нагрузках – быстрая утомляемость. Реже возникают костные деформации.
У ребенка при первичном осмотре могут отмечаться следующие нарушения:
• бледность кожных покровов;
• низкий рост (ниже нормы для конкретного возраста);
• отставание в физическом либо умственном развитии;
• проблемы с зубами – замедленное прорезывание и рост, обширные
поражения кариесом;
• деформация костей: бедренных, лицевой/мозговой части черепа.
Внешний вид пораженной кости на ранних стадиях врожденного остеосклероза остается неизменной. По мере прогрессирования и усугубления патологического процесса проявляются деформации.
Кости становятся ломкими, часто случаются переломы (кости могут ломаться даже от собственного веса).
Однако надкостница при болезни не разрушается, поэтому сращение костей протекает нормально.
Диагностика
Для постановки и уточнения диагноза применяют следующие методы:
• общий анализ крови – позволяет установить наличие анемии, часто сопровождающей мраморную болезнь;
• биохимический анализ крови – проводится для определения содержания в крови ионов фосфора и кальция (их пониженная концентрация говорит о нарушении процессов резорбции костей);
• рентген – при остеопетрозе плотные кости для рентгеновских лучей практически непрозрачны, на снимке представлены темными пятнами, на отдельных участках костей могут быть видны поперечные просветления, костномозговой канал на снимке отсутствует;
• магнитно-резонансная и компьютерная томография – позволяет рассмотреть кость послойно и точно установить степень поражения костной ткани. Наряду с диагностическими методами проводится сбор историй болезни пациента и его близких родственников. Может потребоваться медицинское обследование кого-то из членов семьи.
Профилактика
Высок риск развития остеопетроза у младенца при наличии родственников с данной патологией.
Для определения заболевания у плода применяются методы перинатальной диагностики, позволяющие выявить смертельный мрамор на сроках беременности от 8 недель. Это, скорее, не мера профилактики, а действие, позволяющее супругам принять осознанное решение.
Предупредить патологические изменения в костной ткани невозможно. Можно лишь частично предотвратить переломы и костную деформацию путем оздоровительных процедур (плавание, гимнастика) и правильного питания.
Ранняя форма заболевания характеризуется высоким процентом детской смертности.
Единственным методом, дающим положительные результаты терапии, является трансплантация костного мозга.
Лечение болезни
За последние несколько десятилетий, пересадка костного мозга спасла жизнь не одной сотне ранее обреченных детей. Для трансплантации костного мозга применяется материал, взятый у близкого родственника или донора, подходящего по всем стандартным параметрам, иначе пересаженный костный мозг может не прижиться, и надежды на исцеление не останется. К сожалению, трансплантация костного мозга только препятствует дальнейшему развитию болезни, но не устраняет уже нанесенный вред организму, поэтому проводить ее следует, как можно раньше. В качестве вспомогательной терапии при остеопетрозе, применяются лекарственные препараты и витамины.
• Кальцитриол (активная форма витамина D) – стимулирует остеокласты и
препятствует уплотнению костной ткани.
• Интерферон гамма – обладает одновременно несколькими функциями: стимулирует работу лейкоцитов, повышая, тем самым, иммунитет ребенка;
снижает объем губчатых костей; улучшает показатели крови (гемоглобини и тромбоцитов). Наилучший эффект достигается при комбинации с кальцетонинами.
• Эритропоэтин – применяется для лечения анемии.
• Кортикостероиды – стероидные гормоны, которые препятствуют уплотнению костной ткани и развитию анемии.
• Кальцетонин – пептидный гормон, который в больших дозах улучшает состояние ребенка, однако эффект сохраняется только на время приема препарата.
Немаловажными для укрепления нервно-мышечной ткани и поддержания состояния ребенка являются: физиотерапия и реабилитация, сбалансированная диета, наблюдение у ортопеда, регулярная санация полости рта.
Масштабы заболевания в Чувашии: частота гена среди чувашей - 4,5 % (каждый 1000 брак).
Синдром Дауна
Синдром Дауна – хромосомная аномалия, при которой в кариотипе имеются дополнительные копии генетического материала по 21-ой хромосоме, т. е. наблюдается трисомия по хромосоме 21. Фенотипические признаки синдрома Дауна представлены брахицефалией, плоским лицом и затылком, монголоидным разрезом глазных щелей, эпикантом, кожной складкой на шее, укорочением конечностей, короткопалостью, поперечной ладонной складкой и др. Синдром Дауна у ребенка может быть выявлен пренатально (по данным УЗИ, биопсии ворсин хориона, амниоцентеза, кордоцентеза) или после рождения на основании внешних признаков и генетического исследования. Дети с синдромом Дауна нуждаются в коррекции сопутствующих нарушений развития.
С индром Дауна - аутосомный синдром, при котором кариотип представлен 47 хромосомами за счет дополнительной копии хромосомы 21-ой пары. Синдром Дауна регистрируется с частотой 1 случай на 500-800 новорожденных. Соотношение полов среди детей с синдромом Дауна составляет 1:1. Впервые синдром Дауна описал английский педиатр Л. Даун в 1866 г., однако хромосомная природа и суть патологии (трисомия по хромосоме 21) была выявлена почти столетие спустя. Клиническая симптоматика синдрома Дауна разнообразна: от врожденных пороков развития и отклонений в умственном развитии до вторичного иммунодефицита. Детям с синдромом Дауна требуется дополнительная медицинская помощь со стороны различных специалистов, в связи с чем они составляют особую категорию в педиатрии.
индром Дауна - аутосомный синдром, при котором кариотип представлен 47 хромосомами за счет дополнительной копии хромосомы 21-ой пары. Синдром Дауна регистрируется с частотой 1 случай на 500-800 новорожденных. Соотношение полов среди детей с синдромом Дауна составляет 1:1. Впервые синдром Дауна описал английский педиатр Л. Даун в 1866 г., однако хромосомная природа и суть патологии (трисомия по хромосоме 21) была выявлена почти столетие спустя. Клиническая симптоматика синдрома Дауна разнообразна: от врожденных пороков развития и отклонений в умственном развитии до вторичного иммунодефицита. Детям с синдромом Дауна требуется дополнительная медицинская помощь со стороны различных специалистов, в связи с чем они составляют особую категорию в педиатрии.
В норме клетки человеческого организма содержат по 23 пары хромосом (нормальный женский кариотип 46,XX; мужской - 46,XY). При этом одна из хромосом каждой пары наследуется от матери, а другая – от отца. Генетические механизмы развития синдрома Дауна кроются в количественном нарушении аутосом, когда к 21-ой паре хромосом присоединяется дополнительный генетический материал. Наличие трисомии по 21-ой хромосоме определяет черты, характерные для синдрома Дауна.
Появление дополнительной хромосомы может быть обусловлено генетической случайностью (нерасхождением парных хромосом в овогенезе или сперматогенезе), нарушением клеточного деления уже после оплодотворения либо наследованием генетической мутации от матери или отца. С учетом этих механизмов в генетике различают три варианта аномалии кариотипа при синдроме Дауна: регулярную (простую) трисомию, мозаицизм и несбалансированную транслокацию.
Большинство случаев синдрома Дауна (около 94%) связано с простой трисомией (кариотип 47,XX, 21+ или 47,ХY, 21+). При этом три копии 21-ой хромосомы присутствуют во всех клетках вследствие нарушения разделения парных хромосом во время мейоза в материнской или отцовской половых клетках.
Около 1-2% случаев синдрома Дауна приходится на мозаичную форму, которая обусловлена нарушением митоза только в одной клетке зародыша, находящегося на стадии бластулы или гаструлы. При мозаицизме трисомия 21-ой хромосомы выявляется только в дериватах этой клетки, а остальная часть клеток имеет нормальный хромосомный набор.
Транслокационная форма синдрома Дауна встречается у 4-5% пациентов. В этом случае 21-я хромосома либо ее фрагмент прикрепляется (транслоцируется) к какой-либо из аутосом и при мейозе отходит вместе с ней во вновь образовавшуюся клетку. Наиболее частыми «объектами» транслокации служат хромосомы 14 и 15, реже – на 13, 22, 4 и 5. Такая перестройка хромосом может носить случайный характер или наследоваться от одного из родителей, являющегося носителем сбалансированной транслокации и имеющего нормальный фенотип. Если носителем транслокации выступает отец, то вероятность рождения ребенка с синдромом Дауна составляет 3%, если носительство связано с материнским генетическим материалом, риск возрастает до 10-15%.
Факторы риска рождения детей с синдромом ДаунаРождение ребенка с синдромом Дауна не связано с образом жизни, этнической принадлежностью и регионом проживания родителей. Единственным достоверно установленным фактором, повышающим риск появление ребенка с синдромом Дауна, является возраст матери. Так, если у женщин до 25 лет вероятность рождения больного ребенка составляет 1:1400, к 35 годам уже 1:400, к 40 годам - 1:100; а к 45 - 1:35. Прежде всего, это связано со снижением контроля за процессом деления клеток и увеличением риска нерасхождения хромосом. Однако, поскольку частота родов у молодых женщин в целом выше, то, по статистике, 80% детей с синдромом Дауна рождается от матерей в возрасте до 35 лет. По некоторым данным, возраст отца старше 42-45 лет также увеличивает риск развития синдрома Дауна у ребенка.
Известно, что при наличии синдрома Дауна у одного из однояйцовых близнецов, эта патология в 100% случаев будет иметься у другого. Между тем, у разнояйцовых близнецов, а также братьев и сестер, вероятность такого совпадения ничтожно мала. Среди прочих факторов риска – наличие в роду лиц с синдромом Дауна, возраст матери моложе 18 лет, носительство транслокации одним из супругов, близкородственнные браки, случайные события, нарушающие нормальное развитие половых клеток или зародыша.
Дальтонизм
Дальтонизм – это достаточно распространенное нарушение зрения, которое сопровождается неспособностью глаза воспринимать один или несколько основных цветов. Большинство дальтоников, как правило, не различают какой-то один цвет. Но существует понятие парной слепоты (когда пациенты не видят сразу несколько цветов), а также цветовой слепоты (когда не различают ни одного цвета). Цвета, которые не видят дальтоники, воспринимаются ими, как серый.
История термина
Джон Дальтон был протанопом (не различал красный цвет), но не знал о своей цветовой слепоте до 26 лет. У него были три брата и сестра, и двое из братьев страдали цветослепотой на красный цвет. Дальтон подробно описал свой семейный дефект зрения в небольшой книге. Благодаря его публикации и появилось слово «дальтонизм», которое на долгие годы стало синонимом не только описанной им аномалии зрения в красной области спектра, но и любого нарушения цветового зрения.
Причины дальтонизма
Дальтонизм является наследственным заболеванием, которое вызвано дефектом Х-хромосомы. Но в некоторых случаях заболевание становится следствием нервных или глазных болезней. Независимо от причин, вызвавших данное заболевание, оно, к сожалению, неизлечимо. В силу особенностей генетики, мужчины страдают дальтонизмом гораздо чаще.
Отсутствие цветового восприятия может быть вызвано проблемами функционирования цветочувствительных рецепторов, расположенных в центральной части сетчатки глаза. В качестве рецепторов выступают колбочки (особые нервные клетки).
Различают три вида колбочек, которые отвечают за восприятие основного цвета (красного, зеленого, синего). Люди, в колбочках которых находятся все три типа пигментов, называются трихроматами (имеющие нормальное цветовое восприятие).
Процентное соотношение больных дальтонизмом по России составляет:
30% мужское население из 100%
1% женского населения из 100%
Таблица Рабкина

Таблица Рабкина — что это, принцип действия. Простым диагностическим методом выявления аномального зрения является спектральный способ. Таблицы Рабкина помогают определить и точно дифференцировать три формы отклонения в цветоощущении:
дейтераномалию– нарушение восприятия зеленого спектра;
протаномалию – нарушение восприятия красного спектра;
тританомалия – нарушение восприятия синего.
В каждой из аномалий определяется три степени: А – сильная; В – средняя; С – легкая. При дальтонизме, частичном или полном отсутствии восприятия цветов, тестируемый человек не различает отдельные цвета и видит однородный рисунок. В то время как каждое изображение состоит из большого количества разноцветных кружков и точек одинаковой яркости, но разнящихся по цвету.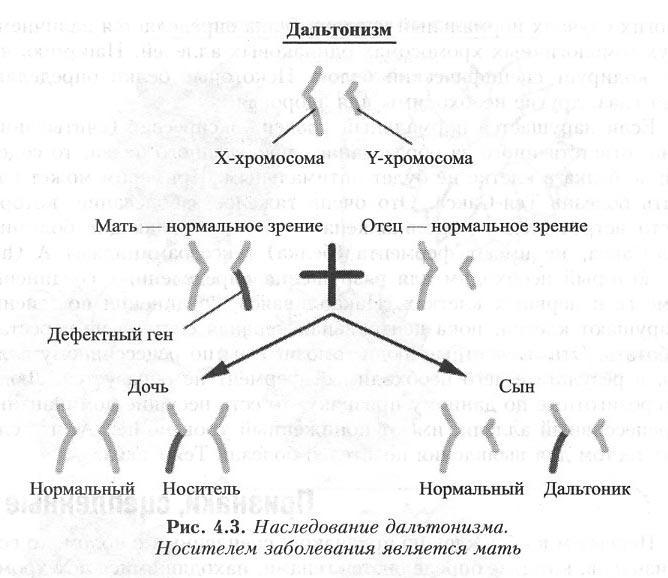
Лечение дальтонизма
Врожденная форма заболевания лечению не поддается. Приобретенная форма заболевания лечится оперативным путем, в зависимости от причин ее возникновения.
Гемофилия
Г емофилия — генетическое, приобретенное по наследству заболевание, которое характеризуется изменением одного гена в Х-хромосоме. Проявлением этого недуга является излишняя кровоточивость и замедленная сворачиваемость крови, так называемая коагуляция. Частота встречаемости в Чувашии (1:22035)
емофилия — генетическое, приобретенное по наследству заболевание, которое характеризуется изменением одного гена в Х-хромосоме. Проявлением этого недуга является излишняя кровоточивость и замедленная сворачиваемость крови, так называемая коагуляция. Частота встречаемости в Чувашии (1:22035)
Этим заболеванием страдают только представители мужского пола. Гемофилия у мужчин появляется в результате наследования болезни от матери. Это значит, что передача заболевания происходит по рецессивному, сцепленному с Х-хромосомой типу. Женщинам при этом недуге отведена роль носителей, или кондукторов. Однако известны редкие случаи, когда заболевали гемофилией и женщины. Это возможно, когда у отца имеется заболевание, а мать является носителем гена гемофилии — дочь таких родителей может родиться с подобным генетическим расстройством.
Одним из заблуждений является утверждение, что человек, страдающий этим генным заболеванием, может умереть от потери крови при любой царапине или порезе. Это не совсем так. Действительно, одним из главных признаков заболевания считают повышенную обильную кровоточивость, но возникает она довольно часто даже в отсутствие травм.
Основными признаками заболевания считаются:
Излишняя кровоточивость, возникающая периодически с различной локализацией: потеря крови при травмах, при удалении зубов, при медицинских вмешательствах, связанных с проведением хирургических операций.
Носовое или десневое кровотечения, которые очень сложно остановить обычными методами. Возможно возникновение самопроизвольного, случайно возникшего кровотечения.
В результате получения легкой, не опасной травмы образуется крупная гематома.
Появление гемартрозов — внутрисуставных кровотечений, являющихся следствием повреждения тканей суставов. Такому явлению обычно сопутствуют острые болевые ощущения, отечность, нарушение двигательной функции сустава. Вторичные гемартрозы могут вести к деформированию сустава и устойчивому нарушению его подвижности.
Проблемы с пищеварением часто сопровождают недуг.
Присутствие крови в моче и кале — опасные симптомы гемофилии. У людей, страдающих этим генетическим заболеванием, достаточно часто встречаются заболевания почек.
Возможно появление таких смертельно опасных признаков как кровоизлияния в головной или спинной мозг.
Причины гемофилии
Гемофилия — недуг наследственный, и поражает, в основном, мужчин. Поскольку ген, отвечающий за гемофилию, расположен в Х-хромосоме, то женщины являются носителями и могут с большой вероятностью передавать заболевание своим сыновьям по наследству. Гемофилия наследуется по рецессивному, сцепленному с Х-хромосомой типу, а так как Х-хромосома у мужчин только одна, то в случае передачи «больной» хромосомы ребенок мужского пола наследует и заболевание.
Врачи могут диагностировать это генетическую аномалию еще до появления ребенка на свет. После рождения явным признаком станут гематомы и излишняя кровоточивость при малозначимых травмах.
О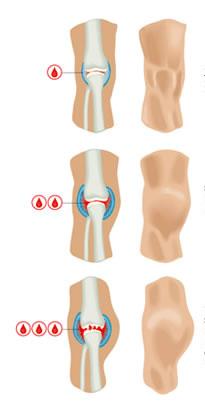 сновные причины гемофилии — наследственные факторы. В настоящий момент медицина не в силах устранить причину возникновения болезни. Пока это невозможно, потому что заболевание оказалось запрограммированным на генетическом уровне. Люди с таким тяжелым недугом должны научиться крайне бережно относиться к своему здоровью, и тщательно соблюдать меры предосторожности.
сновные причины гемофилии — наследственные факторы. В настоящий момент медицина не в силах устранить причину возникновения болезни. Пока это невозможно, потому что заболевание оказалось запрограммированным на генетическом уровне. Люди с таким тяжелым недугом должны научиться крайне бережно относиться к своему здоровью, и тщательно соблюдать меры предосторожности.
Гемофилия бывает трех форм, в зависимости от тяжести заболевания:
Легкая. Кровотечения появляются только после медицинского вмешательства, связанного с проведением хирургической операции, или в итоге полученных травм.
Умеренная. Клинические симптомы, характерные для гемофилии, могут появиться в раннем возрасте. Для такой формы характерно возникновение кровотечений в результате травм, появление обширных гематом.
Тяжелая. Признаки заболевания появляются в первые месяцы жизни ребенка во время роста зубов, в процессе активного движения ребенка при ползании, ходьбе.
Гемофилия у детей до 3 лет может проявляться в виде возникновения гемартрозов. Чаще всего при этом, страдают крупные суставы — тазобедренный, коленные, локтевые, голеностопные, плечевые, лучезапястные. Внутрисуставным кровотечениям сопутствует сильный болевой синдром, нарушения двигательных функций суставов, их отечность, увеличение температуры тела ребенка. Все эти признаки гемофилии должны привлечь внимание родителей.
Типы гемофилииКроме форм тяжести выделяют три подтипа гемофилии:
Гемофилия типа «А» обусловлена генным дефектом, при котором в крови больного отсутствует нужный белок — антигемофильный глобулин, фактор VIII. Такой тип гемофилии называют классическим, и бывает он у 85 процентов всех больных.
Гемофилия типа «В» вызывается недостаточной активностью IX фактора свертываемости крови, при котором происходит нарушение формирования вторичной коагуляционной пробки.
Гемофилия типа «С» вызывается недостатком фактора свертываемости крови XI. Тип С считается самым редко встречающимся.
Типы гемофилии «А», «В» и «С» имеют одинаковые симптомы, но для лечения важным является диагностика типа гемофилии, которая возможна лишь при лабораторном изучении.
Практическая часть проекта
1.1. Решение генетических задач на наследственные заболевания
Задачи с объяснением решения на моногибридное скрещивание
Задача №1
Нормальное здоровье доминирует над признаком синдрома Марфана. Женщина с синдромом Марфана вышла замуж за гетерозиготного здорового мужчину. Какова вероятность рождения детей с синдромом Марфана в этой семье?
(Синдром Марфана - дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии)
Дано: Решение
Человек P: ♀aа x ♂Аа
А-здоровый человек G: а а А а
а-человек с синдромом Марфана F1: 2аа; 2Аа
Найти: F1-?Г-?Ф-? Ф: 2:2
Г: 2:2
Ответ: вероятность рождения детей с синдромом Марфана = 50%
Задача №2
Признак врожденного гипотрихоза доминирует над обычным здоровьем. Гомозиготная женщина вышла замуж за мужчину с врождённым гипотрихозом. Определите какое потомство у них получится?
(Гипотрихоз это заболевание, которое характеризируется полным или частичным отсутствием волос (облысением))
Дано: решение
Человек P: ♀АА x ♂аa
А-человек с врожденным гипотрихозом G А А а а
а -здоровый человек F1:4Aa;
Найти:F1-?
Ф: единообразие
Г: единообразие
Ответ: F1:4Aa
Ф, Г: единообразие
Задача №3
У человека ген врожденной катаракты доминирует над нормальным зрением. У жены зрение нормальное, муж гетерозиготен по гену врожденной катаракты. Определите вероятность рождения в этой семье ребенка с врожденной катарактой.
(Катаракта-помутнение хрусталика глаза)
Дано: решение
Человек Р: ♀аа x ♂Аа
А- ген врожденной катаракты G: а а Аа
а- ген нормального зрения F1: 2Аа:2aa
Найти: F1-? Г-?Ф-? Ф: 2:2
Г: 2:2
Ответ: 50% = вероятность рождения в семье ребенка с катарактой
Задача №4
У человека ген, который не несет развития остеопетроза доминирует над геном, который несет развитие этой болезни. Здоровая женщина у которой отец страдал остеопетрозом, вышла замуж за мужчину, страдающим остеопетрозом.
(Остеопетроз— редкое наследственное заболевание, проявляющееся диффузным уплотнением костей скелета, ломкостью костей, недостаточностью костномозгового кроветворения)
Выясните: 1) Сколько типов гамет образуется у женщины? 2) Сколько типов гамет образуется у мужчины? 3) Какова вероятность рождения в данной семье здорового ребенка 4) Сколько разных генотипов может быть у детей в этой семье? 5) Сколько разных фенотипов может быть у детей в этой семье?
Дано: L-отсутсвие остеопетроза (здоровый)
l -остеопетроз
Фенотип ♀ - отсутсвие остепетроза
Фенотип отца ♀ - остеопетроз
Фенотип ♂ - остеопетроз
Определить: 1)♀n - ?, 2) ♂n - ?, 3) % здоровогоребенка - ?; 4) К-вогенотипов - ?; 5) К-вофенотипов - ?
Решение: Р: ♀Ll x ♂ll
G L l l l
F1 Ll ; ll
F1: генотип – Ll; ll ; Фенотип – здоровые (отсутствие остеопетроза) = 50%; больны остеопетрозом = 50%.
Ответ: 1) два типа; 2) один тип; 3) 50%; 4) два генотипа; 5) два фенотипа.
Задача №5
Ген развития ихтиоза рецессивен по отношению к гену нормального состояния. У здоровых супругов родился ребенок, больной ихтиозом. Определите: 1) Сколько типов гамет может образоваться у отца? 2) Сколько типов гамет может образоваться у матери? 3) Какова вероятность рождения здорового ребенка в данной семье? 4) Сколько разных генотипов может быть у детей в этой семье? 5) Какова вероятность того, что ребенок родится больным?
(Ихтиоз – это генетическое заболевание, при котором нарушается процесс ороговения кожи)
Решение задачи № 5
Дано: D – ген ихтиоза;
d – ген нормального состояния;
Фенотип супругов – здоровы;
Фенотип ребёнка – ихтиозник;
Определить: 1) ♂n - ? 2) ♀n - ? 3) % здорового ребенка ? 4) Количество генотипов ?
5) % второго ребенка (больного)?
Решение: Р: ♂Dd x ♀Dd
♂ D d G D d D d
♀ Dd
F1: генотип – DD : 2Dd : dd
Фнотип - 3 (здоровый) : 1 (ихтиозник)
Ответ: 1) два типа гамет; 2) два типа гамет; 3) 75%; 4) три генотипа; 5) 25%.
Задача №6
Отсутствие олигофрении -доминантный признак. Определить генотипы родителей и детей, Если известно,что мать не болеет олигофренией,а отец болеет олигофренией, з двух детей В семье один здоров другой-болен.
(Олигофрения — это синдром врожденного психического дефекта, выражающегося в умственной отсталости по причине патологии головного мозга)
Дано:
S – отсутствие олигофрении
s – олигофрения
♀ - отсутствие олигофрении
♂ - олигофрения
1(F1) – здоров
2(F1) – болен
Определить: Генотипы Р и F1 - ?
Решение: Р: ♀Ss x ♂ss
G S s s s
F1 генотипы: Ss : ss
Ответ: Р: ♀Ss, ♂ss, (F1) Ss, (F1) ss.
Задача №7
У человека ген полидактилии (многопалости) доминирует над нормальным строением кисти. У жены кисть нормальная, муж гетерозиготен по гену полидактилии. Определите вероятность рождения в этой семье многопалого ребенка.
Решение.
Р: ♀аа х ♂Аа
G: а а А а
F1: Аа, аа, ( 50% на 50%)
Ответ: вероятность рождения многопалого ребенка составляет примерно 50%.
Задача №8
У человека альбинизм – аутосомный рецессивный признак. Мужчина альбинос женился на женщине с нормальной пигментацией. У них родилось двое детей – нормальный и альбинос. Определить генотипы всех указанных членов семьи.
(Альбинизм — это наследственное заболевание, связанное с нарушением пигментного обмена в организме. При этой патологии возникает дефицит меланина — особого вещества, придающего окраску коже, пигментной и радужной оболочкам глаза, волосам, ногтям)
Решение:
Р: ♀Аа x ♂аа
G: А а а а
F1: Аа ;аа
Следовательно, генотип мужа – аа, жены – Аа, ребенка с нормальной пигментацией – Аа, ребенка-альбиноса – аа.
Задача №9
Полидактилия у человека является доминантным признаком, а нормальное строение кистей рук – признак рецессивный. От брака мужчины, имеющего нормальное строение рук с гетерозиготной шестипалой женщиной, родились два ребёнка: пятипалый и шестипалый. Каков генотип этих детей?(Полидактилия – деформация конечности, характеризующаяся наличием дополнительных пальцев на кистях или стопах)
Р: ♀Аа x ♂аа
G: А а а а
F1:Аа (шестипальный); аа (нормальное строение кисти
Ответ: пятипалый ребёнок – рецессивная гомозигота по данному признаку, а шестипалость – гетерозигота.
Задача №10
Ген диабета рецессивен по отношению к гену нормального состояния. У здоровых супругов родился ребенок, больной диабетом. Определите:
1) Сколько типов гамет может образоваться у отца?
2) Сколько типов гамет может образоваться у матери?
3) Какова вероятность рождения здорового ребенка в данной семье?
4) Сколько разных генотипов может быть у детей в этой семье?
5) Какова вероятность того, что второй ребенок родится больным?
(Диабет-болезнь, связанная с обменом веществ, сопровождающаяся обильным выделением мочи и нарушением работы поджелудочной железы)
Дано:
D – ген диабета;
d – ген нормального состояния;
Фенотип супругов – здоровы;
Фенотип ребёнка – диабетик;
Определить:
1) ♂n - ?
2) ♀n - ?
3) % здорового ребёнка - ?
4) К-во генотипов - ?
5) % второго ребёнка (больного) - ?
Решение:
Р: ♂Dd x ♀Dd
G D d D d
F1: генотип – 1DD : 2Dd : 1dd
фенотип - 3 (здоровый) : 1 (диабетик)
Ответ: 1) два типа гамет; 2) два типа гамет; 3) 75%; 4) три генотипа; 5) 25%
Задачи с объяснением решения на дигибридное скрещивание и заболевания, сцепленные с полом
Задача 1.
У отца тугоухость (А) доминирует над нормальным слухом(а), карий цвет глаз (В) – над голубым (b). Запишите генотипы родителей, возможные фенотипы и генотипы детей, родившихся от брака тугоухого голубоглазого мужчины и гетерозиготной кареглазой не глухой женщины.
Тугоухий голубоглазый мужчина aabb.
Гетерозиготная кареглазая не глухая женщина aaBb.
| P | aabb | x | aaBb |
| G | ab |
| aB |
|
|
|
| ab |
| F1 | aaBb | aabb |
|
|
| Нормальный слух. | Нормальный слух. |
|
Задача 2.
У человека катаракта (заболевание глаз) зависит от доминантного аутосомного гена, а ихтиоз (заболевание кожи) – от рецессивного гена, сцепленного с Х-хромосомой. Женщина со здоровыми глазами и с нормальной кожей, отец которой страдал ихтиозом, выходит замуж за мужчину, страдающего катарактой и со здоровой кожей, отец которого не имел этих заболеваний. Составьте схему решения задачи. Определите генотипы родителей, возможные генотипы и фенотипы детей. Какие законы наследственности проявляются в данном случае?
Пояснение.
Отец женщины страдал ихтиозом, значит, Xb она получила от него; отец мужчины не имел катаракты, значит, по первому признаку отец мужчины аа, мужчина соответсвенноАа, т.к. имеет катаркту.
Мама — ааXВXb; папа — AaXВY
Р: ♀ ааXВXb x ♂ AaXВY
G: ♀: aXВ; aXb
♂: AXВ; aXВ; AY ;aY
F1: AaXВXВ — девочка с катарактой и нормальной кожей
АаХВХb — девочка с катарактой и нормальной кожей
ааХВХВ — девочка с нормальным зрением и нормальной кожей
aaXВXb — девочка с нормальным зрением и нормальной кожей
АаХВY — мальчик с катарактой и нормальной кожей
ааХВY — мальчик с нормальным зрением и нормальной кожей
AaXbY — мальчик с катарактой и ихтиозом
aaXbY — мальчик с нормальным зрением и ихтиозом
В этой задаче проявляется закон независимого наследования признаков и признака, сцепленного с полом.
Задача 3.
Мать болеет брахидактилией, тип В имеет волосатый подбородок. Отец здоровый. У них родился ребенок с катарактой и волосатым подбородком. Определите генотипы родителей, первого ребенка, фенотипы и генотипы других возможных потомков. Составьте схему решения задачи. Признаки наследуются независимо.
В потомстве проявились рецессивные признаки, которые у родителей находились в скрытом состоянии.
А - Брахидактилия, тип В, а–здоровый ген.
B –волосатый подбородок , b - гладкий подбородок.
Ребенок aabb, родители A_B_.
Ребенок аа получил одну а от отца, другую от матери; одну b от отца, другую от матери, следовательно, родители AaBb.
|
| AB | Ab | aB | ab |
| AB | AABB | AABb | AaBB | AaBb |
| Ab | AABb | AAbb | AaBb | Aabb |
| aB | AaBB | AaBb | aaBB | aaBb |
| ab | AaBb | Aabb | aaBb | aabb |
9 A_B_ Брахидактилия, тип В,волосатый подбородок
3 A_bbБрахидактилия, тип В, гладкий подбородок
3 aaB_ здоровый,волосатый подбородок
1 aabbздоровый , гладкий побдородок
Задача 4
Отец и мать больны Синдромом Марфана и мать имеет гипотрихоз. От них получены 16 детей: 10 больных и тем и другим, 5 здоровых, но больных гипотрихозом, 3 Марфанабез гипотрихоза, и 2 полностью здоровых. Определите генотипы родителей, потомков и закономерность наследования признаков. Гены двух признаков не сцеплены, доминантные признаки –синдром Марфана (А), гипотрихоз (В).
A - синдром Марфана, а - здоровый.
B - гипотрихоз, b –здоровый.
Отец A_b_, Мать A_B_.
Дети A_B_ 9., aaB_ 3, A_bb 3., aabb1.
Если ребенок имеет аа, то он взял одну а от матери и одну от отца, значит родители AaB_.
Если ребенок имеет bb, то он взял одну b от матери и одну от отца, значит родители AaBb.
|
| AB | Ab | aB | ab |
| AB | AABB | AABb | AaBB | AaBb |
| Ab | AABb | AAbb | AaBb | Aabb |
| aB | AaBB | AaBb | aaBB | aaBb |
| ab | AaBb | Aabb | aaBb | aabb |
9 A_B_ синдром Марфанагипотрихоз
3 A_bbсиндром Марфана без гипотрихоза
3 aaB_ здоровыегипотрихоз
1 aabbполностью здоровые
Закономерность наследования признаков – закон независимого наследования.
Задача 5.
Признаки экстрадактилии доминируют над нормальным здоровьем, а карие глаза над голубыми. Мужчина с экстрадактилией и карие глазами, мать которго была здорова и с голубыми глазами, женился на голубоглазой здоровой жене. Определите генотип и фенотип 1-ого покаления.
Дано:
человек
A –человек с экстрадактилеей
a –здоровый человек
B -кариейглазый
b-голубоглазый
Найти: Ф-? Г-?
Решение:
P ♀aabb x ♂AaBb
GabхAB, AbaB, ab
F1 AaBb; Aabb; aaBb; aabb
Ф: 2:2
Г: 1:1:1:1
ОтветФ: 2:2
Г: 1:1:1:1
Задача 6.
Признак Синдрома Поланда(A) доминирует над нормальным здоровьем(a), а наличие веснушек(B) над отсуствием их(b).Мужчина с генотипом Aabb скрещивается с женщиной имеющей генотип aaBb.Какова вероятность рождения у них ребенка с синдромом Поланда и с веснушками.
Дано:
Человек
A- Синдром Поланда
a-здоровый человек
B-наличие веснушек
b-отсуствие веснушек
Найти: вероятность рождения ребенка с синдромом Поланда и с веснушками
Решение:
P ♀aaBb x ♂Aаbb
G aB, ab хAb, ab
F1 AaBb; Aabb; aaBb; aabb
Ф : 1:1:1:1
Г :1:1:1:1
Ответ: 25%
Задача 7.
Признак синдрома Энжльмена доминирует над нормальным здоровьем, а наличие веснушек над отсуствием их. Гетерозиготная женщина с синдромом Энжльмена и без веснушек скрещивается с здоровым гомозиготным мужчиной с веснушками. Определите фенотип и генотип F1.
Дано:
человек
A-синдром Энжльмена
a-здоровый человек
B-наличие веснушек
b-отсутствие веснушек
Найти:
Решение:
P ♀Aabb x ♂aaBB
G Ab, ab хaB
F1 AaBb; aaBb
Ф: 1:1
Г : 1:1
Ответ: Ф: 1:1
Г : 1:1
Задача 8.
Светлые волосы доминируют над рыжими, а врожденный птоз над нормальным здоровьем. Гомозиготная светловолосая здоровая женщина скрещивает с гетерозиготным рыжим мужчиной с врожденным птозом, мать которго была здорова. Определите какое по фенотипу будетF1.
Дано:
Человек
A-светлые волосы,
a-рыжие волосы
B-Врожденный птоз
b-здоровый человек
Найти:Ф-?
Дано:
P ♀aabb x ♂AaBb
Gab хAB, Ab, aB, ab
F1 AaBb; Aabb; aaBb; aabb
Ф : 1(с.в.):1(с.з.):1(р.в.):1(р.з.)
Г: 1:1:1:1
Ответ:Ф : 1(с.в.):1(с.з.):1(р.в.):1(р.з.)
Заключение
Таким образом, даже сравнительно небольшой перечень наследственных болезней, из установленных на сегодняшний день в Чувашии, указывает на их многочисленность, разнообразие течения и различный прогноз.
Мы рассмотрели некоторые наследственные болезни человека, их классификация, выявлены причины их возникновения, последствия их проявления, методы диагностики, способы лечения. Очень важно вовремя определить (диагностировать) наличие наследственного заболевания у новорождённого, так и предупредить рождение больного ребёнка. С этой целью в стране открыты медико-генетические консультации, в которых врач генетик, даст нужную консультацию. С точки зрения генетики несчастливыми семьями считаются те, в которых есть дети с наследственными болезнями. И самое главное мы должны помнить, что среди нас в нашем обществе есть такие люди, и мы должны толерантно (терпимо) относиться к ним, они тоже имеют право на жизнь.
Огромные шаги по изучению наследственности человека уже сделаны, но у генетики еще очень много проблем впереди, и, возможно, кто-то из вас будет находиться среди тех людей, кто сможет решить проблемы медицины и в том числе проблемы медицинской генетики.
Не забывайте также и то, что наше здоровье во многом зависит еще от правильного образа жизни.
Источники использованной литературы
Биология. Общая биология. Базовый уровень. Учебник 10-11 классы. Беляев, Дымшиц. – М.: Просвещение, 2016.
Л. Берг и С.Н. Давыденков «Наследственность и наследственные болезни человека»
Генетика человека - Учебник - Шевченко В.А., Топорнина Н.А., Стволинская Н.С. – 2002 г.
Н.И. Исаева «О наследственности. Хромосомные болезни человека»
Пренатальная диагностика врожденных пороков развития и наследственных заболеваний, Воронин С.В., Воронина В.Г. – 2008 г
Рубан Э.Д.. Глазные болезни
Ф.А. Самсонов, «Основы генетики и дефектологии»
Н.П. Соколов «Наследственные болезни человека»
Н.Д. Тарасова и Г.Н. Лушанова «Что вы знаете о своей наследственности?»
Интернет ресурсы
Открытая интернет-энциклопедия Википедия "Хромосомные болезни", "Генные болезни"
http://zhenskiy-sait.ru/zdorove/gipotrixoz.html
http://www.vokrugsveta.ru/vs/article/7713/ http://endoinfo.ru/theory_pacients/gipofiz/gipofizarnyy-nanizm- karlikovost.html
https://lecheniedetok.ru/endocrinolog/nanizm.html
http://www.medcentre.com.ua/books/ . http://ozrenii.ru/katarakta/vrozhdennaya.html
https://ru.wikipedia.org/wiki/Альбинизм
https://nebolet.com/bolezni/albinizm.html
https://doctor-neurologist.ru/albinizm-u-cheloveka
https://lookmedbook.ru/disease/albinizm
http://www.rdkb.med.cap.ru/554913/568548/Page.aspx
http://www.krasotaimedicina.ru/diseases/children/haemophilia
https://ru.wikipedia.org/wiki/Гемофилия
https://revolution.allbest.ru/medicine/00881018_0.html
http://www.resp-perinat.med.cap.ru














Вебинар для учителей

Свидетельство об участии БЕСПЛАТНО!
Полезное для учителя

Реализация образовательных программ осуществляется с применением исключительно электронного обучения и ДОТ




Закрыть через 5 секунд
