
Россия, Севастополь


СДЕЛАЙТЕ СВОИ УРОКИ ЕЩЁ ЭФФЕКТИВНЕЕ, А ЖИЗНЬ СВОБОДНЕЕ
Благодаря готовым учебным материалам для работы в классе и дистанционно
Скидки до 50 % на комплекты
только до
Готовые ключевые этапы урока всегда будут у вас под рукой
Организационный момент
Проверка знаний
Объяснение материала
Закрепление изученного
Итоги урока
Была в сети 20.04.2025 18:37

Смирнова Зоя Михайловна
Преподаватель биологии и генетики человека с основами медицинской генетики
67 лет
Местоположение
Специализация
Презентация "Наследственные болезни человека"
Категория:
Биология
16.03.2017 20:49

Просмотр содержимого документа
«Презентация "Наследственные болезни человека"»

НАСЛЕДСТВЕННЫЕ БОЛЕЗНИ ЧЕЛОВЕКА

Актуальность темы
В связи с повышением фона ионизирующей
радиации и загрязнением окружающей среды
мутагенами, количество наследственных
изменений у человека возрастает.
ВОЗ регистрирует ежегодно 3-4 новых
наследственных аномалий. Поэтому
немаловажное значение приобретают знания в области медицинской генетики, основной
задачей которой является выявление и
профилактика наследственных заболеваний.

Наследственные болезни человека –
возникают в результате нарушений в наследственном (генетическом) аппарате половых клеток обоих или одного из родителей.
Рабочая классификация наследственных болезней человека, включает:
- болезни, вызванные мутацией отдельного гена (моногенные или менделевские болезни);
- синдромы, обусловленные хромосомными аномалиями
(хромосомные болезни);
- мультифакториальные заболевания как результат
взаимодействия генетических и средовых факторов (болезни с наследственным предрасположением).
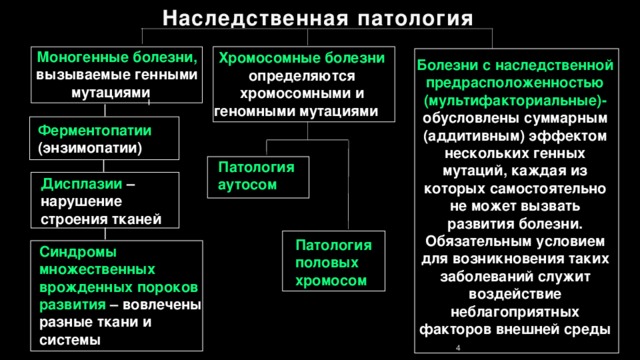
Наследственная патология
Моногенные болезни, вызываемые генными мутациями
Хромосомные болезни
определяются хромосомными и геномными мутациями
Болезни с наследственной предрасположенностью
(мультифакториальные)-
обусловлены суммарным (аддитивным) эффектом нескольких генных мутаций, каждая из которых самостоятельно не может вызвать развития болезни. Обязательным условием для возникновения таких заболеваний служит воздействие неблагоприятных факторов внешней среды
Ферментопатии (энзимопатии)
Патология
aутосом
Дисплазии – нарушение строения тканей
Патология половых
хромосом
Синдромы множественных врожденных пороков развития – вовлечены разные ткани и системы
4

Моногенные болезни –
заболевания в основе которых лежит единичная генная мутация, приводящая к изменению порядка нуклеотидов в ДНК, что влияет на последовательность аминокислот в белке.
Основным признаком, указывающим на моногенный характер патологии,
является менделирующий характер наследования.
До мутации После мутации
Фермент
Сер
Вал
Признак
Н
о
Арг
р
м
а
л
ь
н
ы
й
о
б
м
е
н
РНК Фермент
Ген (ДНК)
Признак
РНК
Т – А
Н
У
Ц
а
Ц – Г
р
Ц
Ц – Г
у
Г
Г – Ц
Т – А
ш
У
Т – А
У
е
Ц
н
Ц – Г
и
Г
Г – Ц
е
Г – Ц
Г
А – Т
А
о
Г – Ц
Г
У
б
Т – А
м
е
н
а
Ген (ДНК)
Т – А
Ц – Г
Г – Ц
Т – А
Т – А
Ц – Г
Г – Ц
Г – Ц
А – Т
Г – Ц
Т – А
У
Ц
Г
У
У
Ц
Г
Г
А
Г
У
выпадение
Сер
Фен
Гли
Сер
5

Болезни аминокислотного обмена –
Фенилкетонурия (ФКУ) – заболевание, обусловленно дефектом фермента фенилаланингидроксилазы, в результате чего нарушается процесс превращения фенилаланина в тирозин.
ФКУ наследуется по А-Р типу.
Частота 1:10000 новорожденных.
В результате дефекта фермента аминокислота
фенилаланин не усваивается организмом.
Неусвоившийся фенилаланин превращается в
фенилпировиноградную кислоту.
Находясь в крови в высокой концентрации,
оказывают токсическое действие на нервные
клетки мозга.
В результате: слабоумие, эпилептические
приступы, расстройство регуляции
двигательных функций.
У больных слабая пигментация вследствие
нарушения синтеза меланина.
А а Х А а
Носители
а
А
а
А
АА А а А а аа
5
больной

Фенилкетонурия (ФКУ)
Диагноз ФКУ ставится простым биохимическим тестом
(проба Феллинга) или микробиологическим тестом Гатри.
Лечение – диетотерапия. Диета исключает мясные, рыбные, молочные
продукты и другие продукты, содержащие животный и, частично,
растительный белок.
Назначают аминокислотные смеси, лишенные фенилаланина
6
Фенилаланин Тирозин

Нарушение обмена углеводов
Галактоземия
- Тип наследования А-Р. Частота 1:50000.
- Болезнь характеризуется поражением ц.н.с, нарушением функции печени, в результате недостаточности фермента галактозо-1-фосфат-уридилтрансферазы.
- Заболевание возникает при вскармливании молоком в результате непереносимости молочного сахара (лактозы), расщепляющегося в кишечнике до галактозы.
- В тканях накапливается избыточное количество продуктов неполного распада лактозы, вызывающих клинические проявления галактоземии у ребенка: рвота, понос, уменьшается масса тела, развивается желтуха и т.д.
В дальнейшем появляются катаракта, цирроз печени, отставание в умственном развитии.
- Диагноз галактоземии ставится на основании обнаружения галактозы в моче.
- Лечение – исключение из пищи молочного сахара.
катаракта
цирроз
печени
в моче
галактоза
6

Наследственные дефекты обмена липидов
Сфинголипидозы – болезни внутриклеточного накопления сфинголипидов, обусловленные дефектом ферментов, катализирующих их расщепление.
Сфинголипиды – структурные компоненты клеточных мембран, в частности миелиновых оболочек нервных волокон
Уоррен Тей–
британский офтальмолог
Болезнь Тея-Сакса
- А-Р тип наследования. Частота 1:50000
- Клиническая картина: поражение ц.н.с. (спинной и головной мозг).
- Интеллект снижается до степени идиотии.
- Двигательные нарушения, приводящие к полной неподвижности.
- Наблюдается снижение зрения, в последующем – атрофия зрительных
нервов и наступает слепота.
- Смерть наступает в 3-4 года.
Бернард Сакс
– американский нейропатолог
15 хромосома генная мутация
9

Болезни стероидного обмена
Адреногенитальный синдром
- А-Р тип наследования.
Частота 1:5000-1:67000.
- Клиническая картина: у девочек заболевание проявляется в форме псевдогермафродитизма, а у мальчиков – преждевременной вирилизацией.
- Синдром обусловлен дисфункцией коры надпочечников (чрезмерная секреция андрогенов). В организме образуется избыток половых гормонов и глюкокортикоидов.
- В моче обнаруживается большие количества андрогенных 17-кетостероидов.
- Исходный пол определяется по половому хроматину в клетках буккального эпителия.
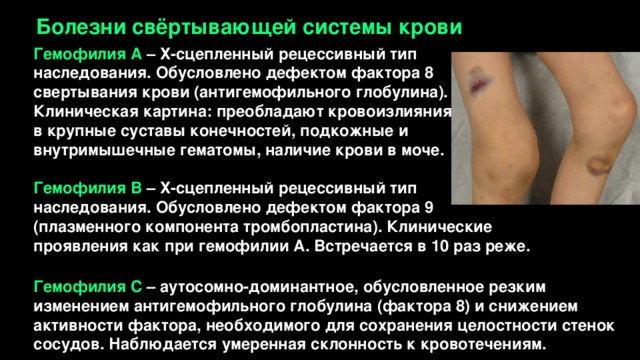
Болезни свёртывающей системы крови
Гемофилия А – Х-сцепленный рецессивный тип наследования. Обусловлено дефектом фактора 8 свертывания крови (антигемофильного глобулина).
Клиническая картина: преобладают кровоизлияния
в крупные суставы конечностей, подкожные и внутримышечные гематомы, наличие крови в моче.
Гемофилия В – Х-сцепленный рецессивный тип наследования. Обусловлено дефектом фактора 9 (плазменного компонента тромбопластина). Клинические проявления как при гемофилии А. Встречается в 10 раз реже.
Гемофилия С – аутосомно-доминантное, обусловленное резким изменением антигемофильного глобулина (фактора 8) и снижением активности фактора, необходимого для сохранения целостности стенок сосудов. Наблюдается умеренная склонность к кровотечениям.

Дисплазии
Синдром Марфана –
наследственная патология соединительной ткани.
А-Д тип наследования; частота 1 : 20000;
Нарушается синтез коллагена и эластина из-за повреждения гена 15 хромосомы, который отвечает за cинтез фибриллина (белок соединительной
ткани, формирующий её эластичность).
- Характерен внешний вид больных:
Патология опорно-двигательного аппарата : длинные и тонкие конечности с такими же пальцами, кифосколиоз, переразгибание в суставах.
Нарушения зрения (подвывих хрусталика, миопия).
Нарушения сердечно-сосудистой системы: поражение клапанов сердца и аневризма аорты.
9

Хромосомные болезни человека, обусловленные изменением структуры
и числа аутосом и половых хромосом
С хромосомными болезнями рождаются менее 1% новорожденных.
Отклонения числа половых хромосом и аутосом связаны с процессом нарушения мейоза. Большинство аномалий несовместимы с жизнью.
Окончательный диагноз хромосомных болезней устанавливается цитогенетическим методом.
Риск рождения ребенка с хромосомными аномалиями увеличивается с возрастом матери.
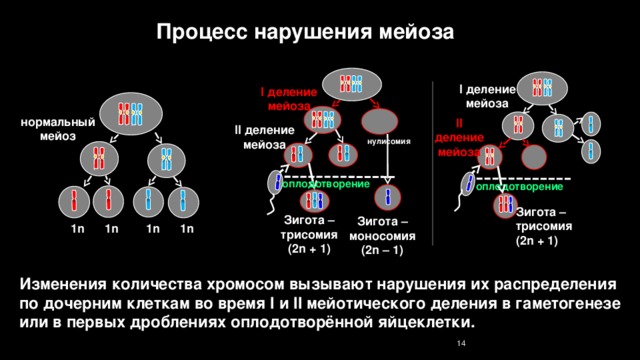
Процесс нарушения мейоза
I деление
мейоза
I деление
мейоза
нормальный мейоз
II деление мейоза
II деление мейоза
нулисомия
оплодотворение
оплодотворение
Зигота – трисомия
(2n + 1)
Зигота – трисомия
(2n + 1)
Зигота – моносомия
(2n – 1)
1n 1n 1n 1n
Изменения количества хромосом вызывают нарушения их распределения по дочерним клеткам во время I и II мейотического деления в гаметогенезе или в первых дроблениях оплодотворённой яйцеклетки.
14

Синдром «кошачьего крика»
- Кариотип 46,XX или ХУ, 5Р- (делеция короткого плеча
пятой хромосомы).
- Частота 1:45000
- Характерно: микроцефалия, умственная отсталость;
- низкая масса при рождении и мышечная гипотония;
- лунообразное лицо с широко расставленными глазами;
- ушные раковины деформированы и низко расположены;
- характерный плач ребёнка, напоминающий кошачье
мяуканье, в результате недоразвития гортани.
- Большинство больных умирают в первые годы,
около 10% больных достигают 10-летнего возраста.
Жером Лежен –
французский ученый
Хромосома 5
15
норма делеция

Синдром Патау
- Кариотип 2n = 47, ХХ+13 – трисомия 13; Частота 1:10000
- Этот синдром представлен двумя вариантами: трисомией
и транслокационной формой: 46, ХХ,-13,-15,+t (q13q15); Клинические признаки:
- выраженная микроцефалия,
- аномалии глазного яблока (микрофтальмия и анофтальм),
- расщепление губы и неба,
- полидактилия,
- врожденные пороки внутренних органов,
- Ранняя смертность, в течение года погибает
- 90% детей. 5% доживают до 3 лет.
Клаус Патау
Трисомия 13 хромосомы
16

Синдром Эдварса
Кариотип 2n=47(+18). Трисомия 18 Частота 1:6500
Клинические признаки:
- выступающий затылок, недоразвитие нижней челюсти,
- деформированные и низко расположенные уши,
- аномалии конечностей, синдактилия.
Патология внутренних органов:
- пороки сердца, гидронефроз, крипторхизм.
Характерна тяжелая задержка умственного развития.
30% погибают в 1 месяц,
менее 10% доживают до года.
Джон Эдварс
Трисомия 18 хромосомы
17

Болезнь Дауна
Кариотип 2n = 47(+21). Трисомия 21.
Возможен и транслокационный вариант:
кариотип 46 хромосом, 14, +t (14,21);
Частота 1:500 - 1:1000
Частота рождения таких детей зависит от возраста матери.
Джон Лэнгдон Даун (1828-1896) английский врач
Транслокационная форма -14, + t (14,21)
(4%)
Трисомия 21
1 2 3 4 5 6 7 8 9
- 2 3 4 5 6 7 8 9
10 11 12 13 14 15 16 17 18
10 11 12 13 14 15 16 17 18
21q
18
19 20 21 22 ху или хх
19 20 21 22 х у х х

Болезнь Дауна
Клинические признаки:
небольшая круглая голова со скошенным затылком, монголоидный разрез глаз, эпикант, короткий нос с широкой плоской переносицей,
маленькие деформированные уши, полуоткрытый рот с высунутым языком, слабоумие. Наблюдаются пороки с.с.с.
Дерматоглифические особенности:
"обезьянья складка" - глубокая поперечная борозда (40%случаев),
единственная сгибательная складка на мизинце (20-25% случаев), складка большого пальца стопы.
- 20-30% погибают до года, 50% - в первые пять лет, 3% доживают до
50 лет.
Эпикантус
Клинодактилия 5-го пальца (искривлённый мизинец) – 60 %
19
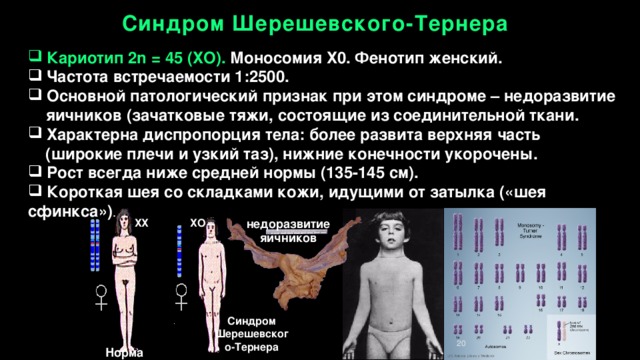
Синдром Шерешевского-Тернера
- Кариотип 2n = 45 (ХО). Моносомия Х0. Фенотип женский.
- Частота встречаемости 1:2500.
- Основной патологический признак при этом синдроме – недоразвитие
яичников (зачатковые тяжи, состоящие из соединительной ткани.
- Характерна диспропорция тела: более развита верхняя часть (широкие плечи и узкий таз), нижние конечности укорочены.
- Рост всегда ниже средней нормы (135-145 см).
- Короткая шея со складками кожи, идущими от затылка («шея сфинкса») .
недоразвитие
яичников
ХХ ХО
Синдром Шерешевского-Тернера
20
Норма

Синдром Шерешевского-Тернера
- Экспресс-диагностика проводится цитологическим методом в
соматических клетках: половой хроматин в клетках у таких
женщин отсутствует.
- Больные бесплодны, т.к. яичники не развиты.
- Введение половых гормонов в период полового созревания,
способствует развитию вторичных половых признаков.
Х- хроматин
У женщин – норма: 46 (ХХ)
ХО
Х- хроматин отсутствует
У женщин – синдром Шерешевского-Тернера: 45(ХО)
21
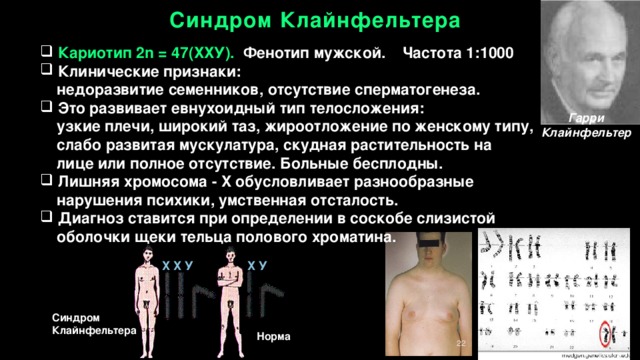
Синдром Клайнфельтера
- Кариотип 2n = 47(ХХУ). Фенотип мужской. Частота 1:1000
- Клинические признаки:
недоразвитие семенников, отсутствие сперматогенеза.
- Это развивает евнухоидный тип телосложения:
узкие плечи, широкий таз, жироотложение по женскому типу, слабо развитая мускулатура, скудная растительность на лице или полное отсутствие. Больные бесплодны.
- Лишняя хромосома - X обусловливает разнообразные
нарушения психики, умственная отсталость.
- Диагноз ставится при определении в соскобе слизистой
оболочки щеки тельца полового хроматина.
Гарри Клайнфельтер
Х Х У Х У
Синдром Клайнфельтера
Норма
22

Другие варианты полисомии половых хромосом
- 47,XXX – трисомия-Х.
Частота 1:1000. Большинство женщин имеют ряд нерезких
отклонений в физическом развитии, нарушения функций
яичников, преждевременный климакс, незначительное
снижение интеллекта. Часто бесплодны, 30% таких больных
сохраняют генеративную функцию.
- 48,XXXX – тяжелое умственное отставание.
- 47,XYY – при увеличении числа У-хромосом половые железы
развиты нормально, рост, как правило высокий, имеются
некоторые аномалии зубов. При этом значительные задержки
умственного развития обнаруживаются редко.
- 48, XXYY, 48,XXXY, 49,XXXYY, 49,XXXXY – другие варианты
синдрома Клайнфельтера. Наблюдаются более глубокие
нарушения физического и психического развития.
22

Аномалии кариотипов при наследственных заболеваниях
Изменение наследственного аппарата
Кариотип
Болезнь
Моносомия по X хромосоме, в том числе и мозаицизм
Синдром Шерешевского - Тернера
45X0;
Синдром Клайнфельтера
Полисомия по X-хромосоме у мужчин
47,XXY; 48,XXXY;
47,ХХ, 13+; 47,ХY, 13+
Трисомия по 13-й хромосоме
Синдром Патау
Синдром Эдвардса
47,ХХ, 18+; 47,ХY, 18+
Трисомия по 18-й хромосоме
47,ХХ, 21+; 47,ХY, 21+
Синдром Дауна
Трисомия по 21-й хромосоме
Делеция короткого плеча
5-й хромосомы
Синдром кошачьего крика
46,XX, 5р- ; 46,ху, 5р-
Делеция короткого плеча
15 хромосомы
Синдром Прадера-Вилли
46 XX или ХУ, 15р-.
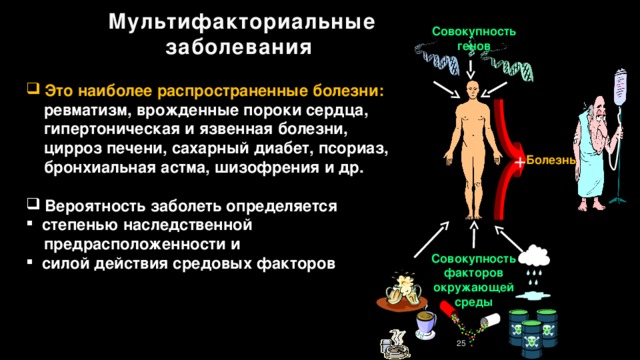
Мультифакториальные заболевания
Совокупность генов
- Это наиболее распространенные болезни:
ревматизм, врожденные пороки сердца,
гипертоническая и язвенная болезни,
цирроз печени, сахарный диабет, псориаз,
бронхиальная астма, шизофрения и др.
- Вероятность заболеть определяется
- степенью наследственной
предрасположенности и
- силой действия средовых факторов
+
Болезнь
Совокупность факторов окружающей среды
25

Лечение наследственных болезней
- Генная терапия –
устранение генетического
дефекта путем введения
генов в клетки пациентов
с целью направленного
изменения генных
дефектов или придания
клеткам новых функций
(например, лечение
врожденного
иммунодефицита в 1990
году при помощи
пересадки гена.
- Предупреждение
заболевания у потомства
(при переносе генов в
половые клетки).
- Патогенетическая
(заместительная,
корригирующая) и
симптоматическая
терапия – нормализация
нарушений без прямого
воздействия на основной
генетический дефект:
- диетотерапия
с исключением поступления
с пищей тех веществ,
концентрация которых в
крови повышена
(например, лечение ФКУ
диетой.)
- заместительная терапия
(гормоны, ферменты и др.
Например, введение
фактора VIII при гемофилии)
- хирургическая коррекция
врожденных пороков и др.
25
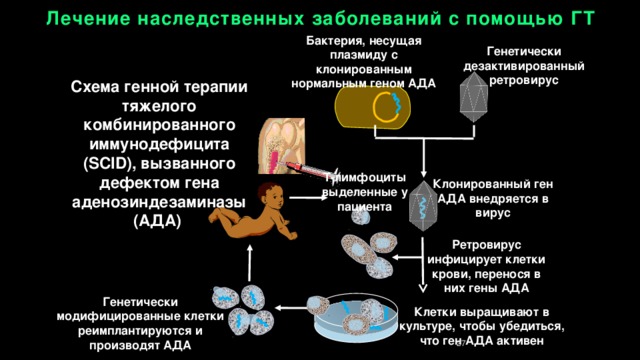
Лечение наследственных заболеваний с помощью ГТ
Бактерия, несущая плазмиду с
клонированным нормальным геном АДА
Генетически дезактивированный ретровирус
Схема генной терапии тяжелого комбинированного иммунодефицита (SCID), вызванного дефектом гена аденозиндезаминазы (АДА)
Т-лимфоциты выделенные у пациента
Клонированный ген АДА внедряется в вирус
Ретровирус инфицирует клетки крови, перенося в них гены АДА
Генетически модифицированные клетки реимплантируются и производят АДА
Клетки выращивают в культуре, чтобы убедиться, что ген АДА активен
25








Вебинар для учителей

Свидетельство об участии БЕСПЛАТНО!

