







© 2025, Сулейманова Умамат Закаржаевна 102 0


СДЕЛАЙТЕ СВОИ УРОКИ ЕЩЁ ЭФФЕКТИВНЕЕ, А ЖИЗНЬ СВОБОДНЕЕ
Благодаря готовым учебным материалам для работы в классе и дистанционно
Скидки до 50 % на комплекты
только до
Готовые ключевые этапы урока всегда будут у вас под рукой
Организационный момент
Проверка знаний
Объяснение материала
Закрепление изученного
Итоги урока
Была в сети 24.11.2025 09:44

Сулейманова Умамат Закаржаевна
преподаватель
Бинарная конференция
Категория:
Биология
19.11.2025 14:22

Просмотр содержимого документа
«Бинарная конференция»

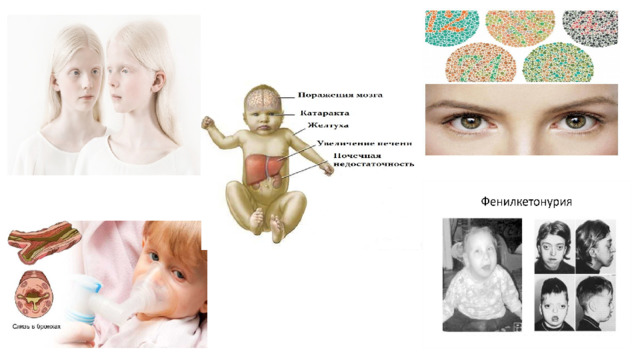
Наследственные генные заболевания. Галактоземия. Муковисцидоз. Фенилкетонурия. Альбинизм.Дальтонизм .

- Генные болезни являются одной из наиболее интересных и
основополагающих проблем в медицинской генетике.
В настоящее время медицинским работникам все чаще приходится сталкиваться с проявлениями наследственной патологии, число которой неуклонно растет. Эти заболевания могут с высокой вероятностью явиться причиной инвалидности или преждевременной гибели пациента.

- Цель конференции:
- изучить основные вопросы этиопатогенеза, основные принципы классификации и диагностики генных болезней, а также разобрать клинические проявления некоторых генных заболеваний.
- Задачи:
- 1. Ознакомиться с этиологией, эпидемиологией и классификацией наиболее часто встречающихся генных заболеваний.
- 2. Разобрать особенности клинических проявлений генных болезней. 3. Изучить общие проблемы лечения и диагностики пациентов с генными болезнями.
Галактоземия.
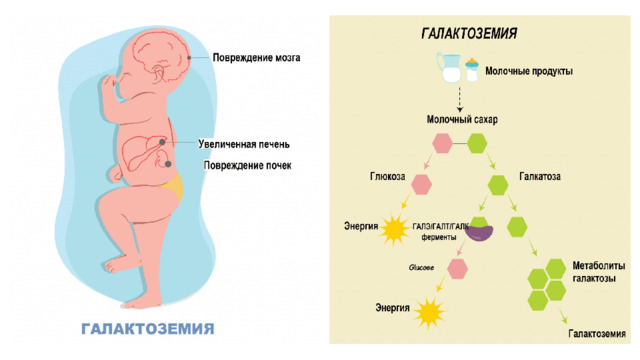
- Галактоземия является наследственным заболеванием, которое характеризуется поражением таких органов, как печень, хрусталик глаза, нервная система. Данная патология связана с нарушением обмена галактозы в человеческом организме.
- Причины галактоземии :
- Известно, что в качестве основной причины заболевания выступает наследственный дефект некоторых генов. Функцией данных генов является контроль над выработкой ферментов, которые осуществляют превращение галактозы в глюкозу. Для галактоземиии характерно накапливание производных галактозы в хрусталике, структурах нервной системы и внутренних органах, где отмечается их токсическое воздействие. Также при данной болезни наблюдается предрасположенность к развитию тяжелых бактериальных инфекций. Это связано с тем, что избыток галактозы подавляет лейкоцитарную функцию.
- Клиническая картина галактоземии
- Возникновение первых симптомов галактоземии происходит через небольшое время после рождения малыша. На фоне кормления молочными продуктами отмечается повторная рвота и нарушение стула (водянистая диарея). В некоторых случаях определяется вздувшийся живот и появление кишечной колики. Отсутствие диагностики данного заболевания у новорожденных чревато увеличением печени и возникновением первых признаков поражения нервной системы. В последнем случае возникают судороги, и отмечается сниженный мышечный тонус. В тяжелых ситуациях происходит формирование цирроза печени.
- Стоит заметить, что для некоторых форм галактоземии характерно медленно прогрессирующее течение. При таких вариантах заболевания отмечается только непереносимость молочных продуктов. Это проявляется спастическими болями в животе, периодической рвотой и диареей. Известно также о бессимптомной форме болезни. Речь идет о галактоземии Дюарте.
- Галактоземия может осложниться циррозом печени, кровоизлиянием в стекловидное тело, катарактой, бактериальным сепсисом и пр.
- Диагностика галактоземии
- Снижение риска возникновения осложнений данного заболевания предусматривает его раннюю диагностику. Некоторые медицинские учреждения обследуют на предмет наличия этого заболевания всех новорожденных.
- Лабораторное определение галактоземии связано с обнаружением увеличенного уровня галактозы в таких биологических жидкостях, как кровь и моча. Чтобы однозначно подтвердить наличие данной болезни, можно воспользоваться генетическим тестированием. Это исследование способствует выявлению мутантного гена, который является причиной развития данного заболевания.
- Оценка контроля болезни и степени поражения внутренних органов предусматривает проведение общего анализа крови и биохимического анализа крови.
- В перечень заболеваний, с которыми необходимо дифференцировать галактоземию, входит врожденная атрезия желчных протоков, сахарный диабет первого типа, прочие нарушения углеводного обмена.
- Лечение галоктоземии
- В основе лечения данного заболевания лежит соблюдение младенцем с первых дней своей жизни безлактозной диеты. Лекарственная терапия галактоземии носит симптоматический характер. Ее целью является оптимизация процессов обмена, а также коррекция функции пораженных органов. Используются препараты, которые улучшают метаболизм. Речь идет об оротате калия и поливитаминах. Вследствие имеющихся в организме расстройств метаболизма, необходимо помнить, что применение лекарственных средств возможно только после консультации с доктором.
- Профилактика галактоземии заключается в генетическом консультировании родителей, которые уже имеют ребенка, страдающего данной патологией.
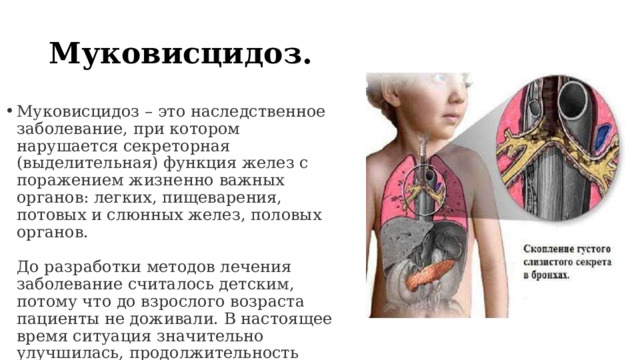
Муковисцидоз.
- Муковисцидоз – это наследственное заболевание, при котором нарушается секреторная (выделительная) функция желез с поражением жизненно важных органов: легких, пищеварения, потовых и слюнных желез, половых органов. До разработки методов лечения заболевание считалось детским, потому что до взрослого возраста пациенты не доживали. В настоящее время ситуация значительно улучшилась, продолжительность жизни пациентов достигает 35 и даже 50 лет. Условие выживания – пожизненный прием лекарств.
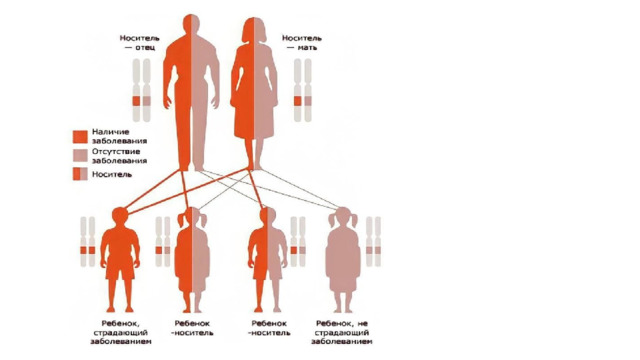
- Особенности муковисцидоза
- Болезнь наследуется по аутосомно-рецессивному типу. Это значит, что оба родителя – носители мутантного (патологически измененного) гена, но заболевание у них не проявилось. Если один родитель здоров, то ребенок тоже будет здоров. Но при слиянии двух патологических генов вероятность рождения больного ребенка составляет 25%. Заболевание выделено в отдельную единицу (нозологию) в 1938 году. Муковисцидоз у детей пытались лечить уже тогда, но успеха не достигли. Во всем мире болеют около 80 тысяч людей. В нашей стране регистрируется один больной на 9 тысяч новорожденных. Российская статистика заболеваемости не отличается от мировой.
- Причины муковисцидоза
- Ответственный за болезнь ген был выделен в 1989 году. Называется он МВТП – муковисцидозный трансмембранный регулятор проводимости, расположен в 7-й хромосоме. Это белок, регулирующий проведение (транспорт) хлора и других электролитов сквозь мембраны клеток эпителия, которым покрыты выводящие протоки желез внутренней секреции. Из-за мутации структура этого белка нарушается, и секрет (отделяемое) желез приобретает вязкость и ненужную густоту. Поражаются такие органы:
- бронхи и легкие;
- печень;
- поджелудочная железа;
- кишечник;
- репродуктивные органы;
- слюнные, слезные и потовые железы.
- Густое отделяемое не может покинуть выводной проток, застаивается, отчего протоки постепенно расширяются. В первую очередь страдают органы дыхания и пищеварения, в расширенных протоках образуются мелкие кисты. Поскольку секрет плохо выводится, железа постепенно снижает свою активность, медленно атрофируется, железистая ткань погибает и замещается соединительной. На этом этапе появляется специфический признак муковисцидоза: соленый привкус кожи (под микроскопом видны кристаллы соли). Патологическому процессу более всего подвержены легкие: застой слизи провоцирует хроническое воспаление, расширенные железы бронхов перекрывают просвет, образуются бронхоэктазы или хронический нагноительный процесс в местах расширений. Некоторые участки легкого спадаются, образуя очаги ателектаза. Легочная ткань перерождается, заменяется фиброзной или плотной соединительной тканью. Аналогичные процессы проходят во всех органах, где есть железы внутренней секреции.
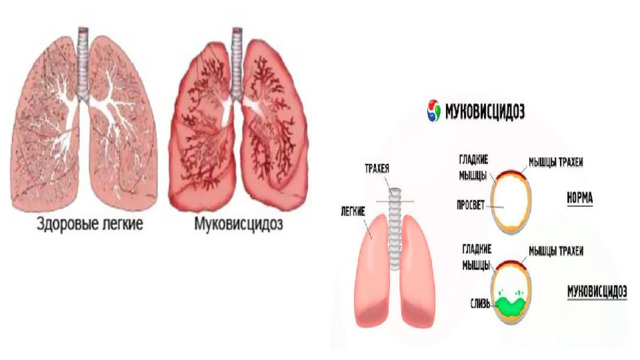
- Симптомы муковисцидоза
- Тяжесть заболевания во многом зависит от возраста, в котором обнаружились первые проявления. Меньше всего шансов имеют новорожденные и дети первого года жизни, даже несмотря на проводимое лечение. Для всех возрастов характерны легочные и желудочно-кишечные проявления: одышка, шумное со свистом дыхание, плохой набор веса, частые поносы. Муковисцидоз у новорожденных проявляется мекониевой (меконий – первородный кал) непроходимостью кишечника, ребенок не выделяет кал. В первые сутки развивается обструктивная (вследствие закупорки общего желчного протока) желтуха. Бывает частая рвота желчью, на животе появляется рисунок сосудов. Младенец страдает от малокровия или анемии . Выпячивается большой родничок, часто бывает асимметрия лица из-за паралича лицевого нерва. Младенец очень плохо набирает вес, значительно отстает от сверстников в физическом развитии. Если опустить ладошку в воду, то пальчики быстро сморщиваются – вода растворяет соли на коже. Такие дети плохо переносят смеси для искусственного вскармливания. Муковисцидоз у детей постарше проявляется синдромом обструкции (сужения просвета) бронхиального дерева. Беспокоят удушье на вдохе, удлиненный выдох. У детей увеличиваются печень и селезенка, воспаляются придаточные пазухи носа, они не переносят жару. Нарушается углеводный обмен, формируется цирроз печени, дети пьют много воды и выделяют много мочи. Характерен симптом «барабанных палочек» – утолщение концевых фаланг стоп и кистей. Становится заметным отставание в физическом и нервно-психическом развитии.
- Диагностика муковисцидоза
- Диагностика начинается в роддоме, где всем новорожденным проводят скрининг, начиная с четвертого дня жизни. Определяют уровень ИРТ – иммунореактивного трипсина (кровь из пятки наносят на фильтровальную бумагу, которую затем исследуют в лаборатории). Если показатель выше нормы, повторяют исследование на 28-й день. При повторном показателе выше нормы проводят потовую пробу (определяют количество натрия и хлора на коже), обследование на сахарный диабет первого типа и рентгенографию кишечника. Основной анализ на муковисцидоз – генетический, но точность его составляет не более 90%. У патологического гена около 2 тысяч мутаций, в то время как лаборатории располагают возможностью определения не более 30.
- Лечение муковисцидоза
- Применяется симптоматическая терапия, поскольку вылечить это заболевание невозможно. Лечение – пожизненное, направлено на компенсацию функций пострадавших органов. Питание должно содержать много животного белка – мясо, рыбу, яйца, молочные продукты (если нет лактазной недостаточности). Больному требуется много жидкости, которую нужно подсаливать. Для улучшения функции легких нужна лечебная гимнастика, используются положения тела, при котором грудная клетка растягивается. Полезно громко кричать, петь, постукивать рукой по грудной клетке. Используют особое устройство – виброжилет, имитирующий движения грудной клетки при кашле.

Фенилкетонурия.
- Фенилкетонурия (ФКУ) — это генетическое заболевание, которое приводит к накоплению аминокислоты фенилаланина в организме из-за врожденной недостаточности фермента фенилаланингидроксилазы. Его важно своевременно выявить, так как нелеченая патология нарушает развитие мозга ребенка, провоцирует снижение интеллекта и психические расстройства.
- Причины фенилкетонурии у детей
- Известно более тысячи мутаций, которые вызывают фенилкетонурию. Обычно болезнь связана с мутацией гена PAH. В результате понижается активность фермента фенилаланингидроксилазы (ФАГ).
- В норме фенилаланин постоянно утилизируется за счет превращения в тирозин . В свою очередь, из аминокислоты тирозина затем образуются биогенные амины (биологически активные низкомолекулярные белковые вещества) и пигмент меланин. Небольшое количество фенилаланина также используется для производства белковых молекул.
- За превращение фенилаланина в тирозин отвечает фермент ФАГ. Если его в организме не хватает, то это влечет два последствия:
- фенилаланина становится все больше, потому что он не утилизируется;
- развивается дефицит тирозина.
- Когда уровень фенилаланина повышается слишком сильно, начинается его превращение в другие вещества: метаболиты фенилаланина (фенилпируват, фенилацетат, фениллактат и другие). Они оказывают токсическое воздействие на организм.
- С другой стороны, дефицит тирозина приводит к нехватке веществ, которые из него образуются: миелин (нужен для оболочек нервов), серотонин и норадреналин (важны для работы нервной системы и созревания мозга), меланин (пигмент, окрашивающий кожу).

- Классификация
- Фенилкетонурия относится к гиперфенилаланинемиям (состояниям с повышенным уровнем фенилаланина) . Их делят на два типа:
- ФАГ-зависимая — связанная с дефицитом фермента ФАГ (по МКБ-10 классифицируется как E70.0 — классическая фенилкетонурия);
- ФАГ-независимая — связанная с нарушением биосинтеза или восстановления тетрагидробиоптерина (кофермент), который необходим для фермента ФАГ (E70.1 — другие гиперфенилаланинемии).
- По максимальной концентрации фенилаланина в крови на момент выявления фенилкетонурии у новорожденных болезнь классифицируют по степеням тяжести: легкая, средняя или тяжелая.

- Симптомы :
- Ребенок с фенилкетонурией рождается здоровым, без симптомов. Они проявляются спустя несколько месяцев.
- Основные признаки фенилкетонурии у ребенка без лечения:
- вялость или беспокойство; отсутствие интереса ко всему происходящему;
- повышенный тонус мышц; сыпь на коже;
- при объективном обследовании определяются усиленные рефлексы.
- При фенилкетонурии появляется запах, который называют «мышиным» . Он связан с выделением метаболитов фенилаланина через пот и мочу.
- Если после рождения ребенка с фенилкетонурией болезнь не диагностировали, а лечение не назначили, то после полугода появляются такие симптомы у грудничков:
- отсутствие реакции на речь;
- ребенок не узнает людей; нет фиксации взгляда;
- нет интереса к игрушкам; ребенок не сидит, не переворачивается на живот.

При объективном обследовании врачи определяют уменьшение окружности головы. Зубы у ребенка прорезываются поздно. Кожа и волосы обычно светлые, глаза – голубые, что связано с дефицитом пигмента меланина.
Обычно фенилкетонурия у детей вовремя выявляется и лечится. Но даже у пациентов, которым назначено лечение, возможны такие симптомы, как:
плохая концентрация внимания;
замедленное речевое развитие;
плохая память.
У многих встречаются пограничные психические расстройства, чрезмерная тревожность, эмоциональная неустойчивость, агрессивность.
- Верхней нормой фенилаланина в крови считается 120 мкмоль/л . Если уровень выше, то дальнейшая диагностика включает повторное определение фенилаланина, а также тирозина и соотношения этих двух веществ. Высокую вероятность ФКУ констатируют при соотношении более 3, в то время как в норме оно должно быть менее 1. Для полной потери активности фермента характерен уровень фенилаланина более 1200 мкмоль/л. Это тяжелая форма заболевания, которую также называют классической. Для подтверждения диагноза проводят генетические исследования. Они позволяют выявить мутации в генах. При неоднозначных результатах генетических анализов определяют птерины (кофактор для фермента ФАГ) в моче. Для оценки потенциального ответа на кофакторную терапию проводят нагрузочный тест с препаратом сапроптерином в течение двух суток. При наличии неврологических симптомов потребуется МРТ мозга. С трех лет можно также проводить КТ мозга для выявления отложения солей кальция (кальцификатов) в структурах полушарий головного мозга (базальных ганглиях). Для выявления эпилептической активности мозга проводят ЭЭГ. По показаниям делают УЗИ брюшной полости, ЭКГ, эхокардиографию. Детям старше пяти лет проводят денситометрию — рентгенологическое исследование для оценки минеральной плотности костной ткани.
- Диагностика
- Обычно ФКУ у новорожденных обнаруживают своевременно за счет скрининга: при рождении у малышей берут кровь на фенилаланин.
- Если скрининг не проводили, то поводом для назначения этого анализа может быть:
- эпилепсия;
- умственная отсталость;
- «мышиный» запах;
- аутизм;
- тремор;
- первый фототип кожи (светлая кожа).
- Лечение фенилкетонурии у детей
- Тактика ведения пациентов отличается в зависимости от степени тяжести заболевания.
- Легкая форма. Не всем детям, которые больны ФКУ, нужно лечение: при легкой форме симптомы фенилкетонурии отсутствуют, а существенных нарушений метаболизма нет. Таким пациентам потребуется лишь наблюдение и периодический контроль уровня фенилаланина в крови.
- Умеренная форма. При этой степени тяжести заболевания ФКУ у детей частично сохраняется активность фермента ФАГ. Таким пациентам назначают диету . Потребность в медикаментозной терапии зависит от результатов теста на чувствительность к аналогу тетрагидробиоптерина.
- Тяжелая форма. Активность фермента ФАГ минимальная. Ребенку понадобится строгая диета и, возможно, кофакторная терапия.

Альбинизм.
- Альбинизм (от лат. albus — белый) рассматривается как наследственное заболевание и характеризуется врожденным частичным или полным отсутствием в организме пигмента меланина. Таким образом, кожа, волосы и радужная оболочка глаз выглядят обесцвеченными, бледными или имеют более светлый оттенок, чем должны были быть в норме.

- Причины появления альбинизма Цвет кожи детерминирован генетически. Наличие альбинизма говорит о мутации генов (на настоящий момент описаны 19 генов, отвечающих за появление альбинизма), ответственных за цвет кожи, волос и радужной оболочки глаз. Эти мутации приводят к недостаточной активности фермента тирозиназы, благодаря которой синтезируется аминокислота тирозин, являющаяся основным регулятором производства пигмента меланин – так схематично и очень упрощенно выглядит меланогенез. Заболевание передается по аутосомно-рецессивному (то есть оба родителя должны являться носителями рецессивного гена) и Х-сцепленному (только в случае с геном GRP143) типам наследования.
- Классификация заболевания
- Ранее случаи альбинизма разделяли только по внешним (фенотипическим) проявлениям – полный и неполный.
- К первому относились все типы альбинизма с выраженными нарушениями пигментации глаз, кожи и ее придатков. К неполным формам – глазные типы заболевания, когда отмечался недостаток пигментации радужной оболочки, а также те разновидности патологии, которые приводили к пятнистости кожи. В настоящее время принята классификация, основанная на генетических аспектах: Глазокожный альбинизм тип 1А – полное отсутствие пигмента меланина в организме. Глазокожный альбинизм тип 1В – при этом типе синтезируется дефектный фермент тирозиназа, поэтому синтез меланина возможен, но с имеет разную степень выраженности.
Температурно-чувствительный глазокожный альбинизм – активность синтеза пигмента зависит от температуры тела. Для него характерно падение активности фермента тирозиназы, если температура тела более 37̊С. При этом типе альбинизма преобладают поражения глаз, поскольку их температура всегда выше 37̊С. Глазокожный альбинизм тип 2 – активность фермента тирозиназы, нормальная, но нарушен транспорт тирозина, в результате чего меланин не синтезируется. Глазокожный альбинизм тип 3 – встречается только у африканцев, формируется коричневая окраска кожного покрова. Глазной альбинизм рецессивный и связанный с Х-хромосомой – синтез пигмента частично нарушен. Аутосомно-рецессивный глазной альбинизм – данный тип не удалось связать с какими-либо генетическими нарушениями. Кроме изолированных глазного и глазокожного типов существуют редкие наследственные синдромы альбинизма, которые связаны с системными поражениями. К ним относятся синдром Германски-Пудлака и синдром Чедиака-Хигаси, встречающиеся, к счастью, крайне редко.
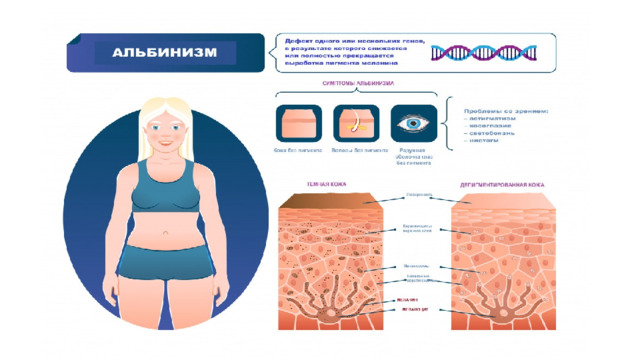

- Симптомы альбинизма Признаки альбинизма, как правило, очевидны, однако могут быть выражены слабо (это зависит от типа альбинизма и дефектного гена). При видимых нарушениях обесцвечены и кожа, и волосы, и радужная оболочка глаз. Один из важных симптомов альбинизма – зрительные нарушения:
- нистагм – быстрые, непроизвольные возвратно-поступательные движения глаз;
- косоглазие – неспособность обоих глаз оставаться направленными в одну и ту же точку или двигаться в унисон;
- экстремальная близорукость или дальнозоркость;
- светобоязнь – чувствительность к свету, иногда переходящая в дневную слепоту;
- астигматизм – аномальная кривизна передней поверхности глаза или внутренней части глаза, вызывающая снижение зрительной функции.

- Диагностика альбинизма Диагноз ставится сразу после рождения ребенка. Врач-педиатр оценивает состояние пигментации кожи и волос, а офтальмолог – наличие той или иной зрительной патологии. Подтвердить диагноз альбинизма может врач-генетик, проведя секвенирование генов. Очень важно выявить наследственный характер патологии и собрать анамнез. Редким и дорогостоящим методом, но позволяющим уточнить прогноз заболевания, является определение тирозиназной активности в тканях (например, в волосяных фолликулах).
- Чем лучше сохранена активность тирозиназы в тканях, тем ниже выраженность других симптомов альбинизма.
- Лечение альбинизма На сегодняшний день специфического лечения альбинизма не существует, поэтому усилия врачей направлены на коррекцию остроты зрения больных и профилактику злокачественных патологий кожи.
- Осложнения
- Из-за отсутствия меланина, выполняющего защитную функцию, люди с альбинизмом имеют высокий риск рака кожи.
- Со стороны органов зрения: снижение остроты зрения, отслойка сетчатки.

Дальтонизм.
- Дальтонизм – нарушение, когда человек не различает часть цветов солнечного спектра. Диапазон оттенков у него значительно уже, чем у людей с нормальным зрением.
- Патология названа по фамилии врача Джона Дальтона, жившего в 18 столетии: у него самого была такая аномалия. Дальтоники не видят один или два цвета из трех - красный, зеленый или синий. У некоторых наблюдается тяжелая степень нарушения: они не различают все три цвета.
- Дальтонизм выявляется в основном у мужчин: пять-девять процентов сильной половины человечества сталкиваются с такой проблемой. У женщин встречается намного реже – около 0,4-0,5 процента. Среди дальтоников немало известных людей, в том числе художники Ван Гог и Михаил Врубель.
Классификация :
- В соответствии с тем, какие цвета не может различать человек, выделяют типы дальтонизма.
- Аномальная трихромазия. В норме человеческий глаз распознает три основных цвета – синий, красный и зеленый. Из них складываются другие оттенки. Когда цветовое восприятие искажено, говорят об аномальной трихромазии. Человек различает оттенки, но не так ярко и интенсивно, как в норме. Восприятие красного снижено при протаномалии (протанопии). Зеленого – при дейтераномалии (дейтеранопии). Синего – при тританомалии (тританопии).
- Ахромазия характеризуется невозможностью воспринимать все три цвета. Окружающее предстает в черно-бело-серых тонах. Это происходит потому, что из всех фоторецепторов в глазах есть только те, что воспринимают эти цвета. Патология диагностируется у одного человека на триста тысяч.
- Монохромазия – аномалия, при которой доступно восприятие только одной гаммы. Если это желтый, говорят о ксантопсии, синий – цианопсии, зеленый – хлоропсии, красный – эритропсии. Часто у дальтоника с монохромазией наблюдаются увеличение светочувствительности либо светобоязнь, нистагм – тремор глазных яблок.
- Дихромазия означает, что воспринимается лишь два цвета из трех основных. Когда невозможно видеть красный, диагностируется протанопия: он кажется темно-зеленым, коричневым, серым или черным. Синий и фиолетовый – тританопия. Зеленый – дейтеранопия: вместо него видится розоватый либо бледно-оранжевый.
- В соответствии с тем, насколько выражены проявления, выделяют три степени дальтонизма. А – максимальное нарушение. В – патология средней тяжести. С – легкая форма.

Причины развития дальтонизма
- Патология возникает, когда светочувствительные рецепторы сетчатки глаза работают неправильно. Колбочки и палочки содержат пигменты, чувствительные к цветам. Благодаря им люди видят разные оттенки. Палочки «отвечают» за восприятие черно-белых тонов, а колбочки – красного, зеленого и синего, из которых получаются другие цвета. Нормальный глаз человека может различать около полутора тысяч оттенков.
- В случае развития дальтонизма колбочки функционируют неправильно. При отсутствии в них одного из пигментов (белков) они не «видят» цвета какого-то спектра. Когда нужного белка мало, оттенки кажутся неяркими, темными, серыми. Иногда пигменты находятся не в тех участках сетчатки, где положено. Тогда одни оттенки воспринимаются как другие.
- Чаще всего нарушение является наследственным. Обычно мутантный ген находится в Х-хромосоме и передается по мужской линии. Носителем гена могут быть представители обоих полов.
- Намного реже дальтонизм приобретается в течение жизни. Это происходит из-за травм сетчатки и зрительного нерва; помутнения хрусталика с возрастом, воздействия некоторых лекарств, нейродегенеративных заболеваний, повреждений затылочной доли головного мозга, болезни Паркинсона, инсульта.

Симптомы дальтонизма у детей
- Дети с дефектным геном страдают дальтонизмом с самого рождения. Однако родители могут долго этого не замечать. Когда малыш подрастает, дальтонизм у ребенка можно заподозрить по таким признакам.
- Ребенок не может верно назвать цвета предметов.
- Рисует, выбирая для изображения объектов цвета, которые нетипичны для них.
- Вместо оранжевого ему видится зеленый, а темно-зеленый воспринимается как бордовый.
- Не различает пастельных тонов: они ему кажутся белыми.
- Диагностируется нарушение обычно в школе во время плановых осмотров, иногда – уже во взрослом возрасте.

Диагностика дальтонизма Для определения патологии врачи-офтальмологи используют специальные полихроматические таблицы. На листах нарисованы разноцветные точки и кружочки, которые складываются в цифры. Дальтоникам не удается их распознать: они видят лишь цветные круги. При диагностике маленьких детей применяется электроретинография, которая помогает понять, насколько активна сетчатка. Освещая глаз красными или синими лучами, врач отслеживает электрические импульсы в ней. Может использоваться способ Гольмгрена. Пациенту предлагается разложить цветные нитки по трем группам спектра. Врач также проводит исследования, используя специальные приборы: спектраномалоскоп, аномалоскоп и другие. Применяет периметрию, рефрактометрию, биомикроскопию, офтальмоскопию, УЗИ и так далее.

Методы лечения дальтонизма.
- Врожденный дальтонизм – патология, которая не поддается полному излечению. Приобретенный в определенных случаях можно исцелить, если выявлена причина развития нарушения. Когда дальтонизм вызван помутнением хрусталика глаза, решить проблему помогает его замена на искусственный. Если причина в макулярном диабетическом отеке, применяется лекарственная терапия.
- Скорректировать цветовое восприятие помогают специальные очки и линзы пяти разных типов.
Вебинар для учителей

Свидетельство об участии БЕСПЛАТНО!

