
Россия, Омск


СДЕЛАЙТЕ СВОИ УРОКИ ЕЩЁ ЭФФЕКТИВНЕЕ, А ЖИЗНЬ СВОБОДНЕЕ
Благодаря готовым учебным материалам для работы в классе и дистанционно
Скидки до 50 % на комплекты
только до
Готовые ключевые этапы урока всегда будут у вас под рукой
Организационный момент
Проверка знаний
Объяснение материала
Закрепление изученного
Итоги урока
Была в сети 27.03.2026 09:53

Грудецкая Виктория Ивановна
Преподаватель биологии, химии, экологии
36 лет
Местоположение
Специализация
Наследственные болезни человека, их причины и профилактика
Категория:
Биология
24.04.2017 11:24

Просмотр содержимого документа
«Наследственные болезни человека, их причины и профилактика»
Наследственные болезни человека, их причины и профилактика

Наследственные болезни –
заболевания человека, обусловленные хромосомными и генными мутациями
Выделяют три основные группы наследственных заболеваний:
1) Хромосомные болезни
2) Генные (молекулярные) болезни
3) Мультифакториальные заболевания

1. Хромосомные болезни
- Хромосомные болезни – наследственные заболевания, которые обусловлены геномными (изменение числа хромосом) и хромосомными (изменение структуры хромосом) мутациями
- Основная причина возникновения хромосомных болезней – нерасхождение хромосом в мейозе во время гаметогенеза у одного из родителей
- Они возникают вследствие мутаций в гаметах одного из здоровых родителей или в зиготе на первых стадиях дробления

Хромосомные болезни:
- Синдром Дауна
- Синдром Клайнфельтера
- Синдром трисомии
- Синдром Эдвардса
- Синдром Орбели
- Синдром Патау
- Синдром Шерешевского-Тернера

Синдром Дауна
- Синдром Дауна - аутосомный синдром, при котором кариотип представлен 47 хромосомами за счет дополнительной копии хромосомы 21-ой пары
- Впервые синдром Дауна описал английский педиатр Джон Лангдон Даун в 1866 г.
- Кариотипы больных – 47, ХХ, 21+ или 47, ХУ, 21+
- Частота: 1 случай на 500-800 новорожденных
- Соотношение полов среди детей с синдромом Дауна составляет 1:1
- Клиническая симптоматика синдрома Дауна разнообразна: от врожденных пороков развития и отклонений в умственном развитии до вторичного иммунодефицита

Синдром Дауна

Синдром Клайнфельтера
- Синдром Клайнфельтера – это генетическое заболевание, встречающееся только у мужчин и характеризующееся наличием в кариотипе одной или нескольких дополнительных Х-хромосом
- Синдром был впервые открыт в 1942 году доктором Гарри Клайнфельтером
- Кариотип 47, ХХУ . Частота 1:400
- Клинические признаки: высокий рост, длинные конечности, гинекомастия (увеличения молочных желез), отсутствие сперматогенеза, недоразвитие половых желез
- Иногда больные имеют 48 и 49 хромосом (48, ХХXY; 49, ХХХХY)
- Чем больше Х-хромосом в кариотипе, тем высшая вероятность развития умственной отсталости

Синдром Клайнфельтера

Трисомия по Х-хромосоме
- Трисомия по Х-хромосоме ( 47,ХХХ ) встречается у новорожденных девочек с частотой 1:1000
- Редко диагностируется в раннем детстве; взрослые больные обычно имеют нормальный женский фенотип
- Немногочисленные исследования показали, что у женщин с кариотипом 47,ХХХ наиболее часто отмечаются: высокий рост, умственная отсталость (как правило, легкой степени), позднее развитие речи, эпилепсия, бесплодие
- Риск рождения ребенка с трисомией по Х-хромосоме повышен у пожилых матерей
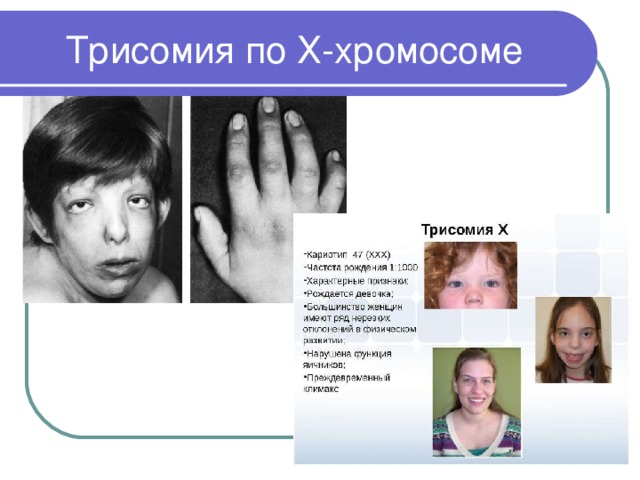
Трисомия по Х-хромосоме
Синдром «кошачьего крика»
- Синдром «кошачьего крика» (синдром Лежена) – редкое хромосомное заболевание, при котором у больных наблюдается дефект в строении пятой хромосомы. Данный дефект сопровождается множественными аномалиями развития различных органов и тканей
- Частота: 1:40000 - 1:50000 новорожденных
- Признаком его служит необычный плач детей, напоминающий мяуканье или крик кошки. Это связано с патологией гортани или голосовых связок. Однако с возрастом этот крик исчезает.
- Наиболее типичным, помимо "кошачьего крика", является умственное и физическое недоразвитие, микроцефалия (аномально уменьшенная голова)
- Своеобразен внешний вид больных: лунообразное лицо, микрогения (маленькие размеры верхней челюсти), эпикант (вертикальная складка кожи у внутреннего угла глазной щели), высокое небо, плоская спинка носа, косоглазие. Ушные раковины расположены низко и деформированы, врожденные пороки сердца, патология костно-мышечной системы, синдактилия стоп (полное или частичное сращение соседних пальцев), плоскостопие, косолапость и др.), мышечная гипотония. Большинство детей умирает в раннем возрасте
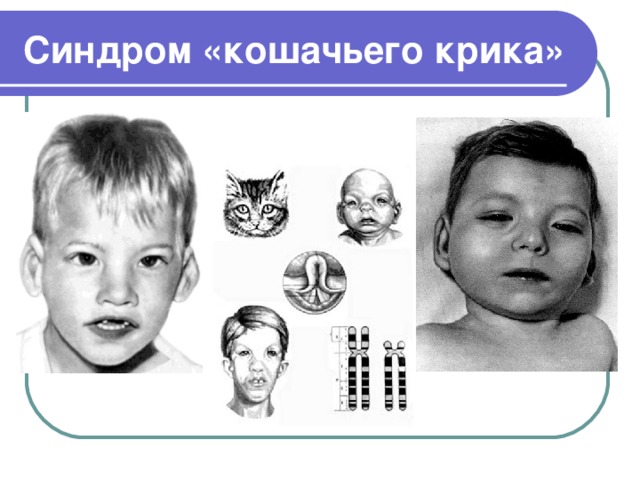
Синдром «кошачьего крика»

Синдром Эдвардса
- Синдром Эдвардса является одной из форм редкого генетического заболевания, когда часть 18-хромосомы человека дублируется. Кариотип 47, ХХ, 18+ или 47, ХУ, 18+
- Большинство детей с данной патологией умирают еще на стадии эмбрионального развития, это происходит в 60 % случаев
- Распространенность синдрома Эдвардса в среднем составляет 1:3000 - 1:8000 случаев
- Синдром Эдвардса был назван в честь доктора Джона Эдварда, который в 1960 году описал первые случаи и зафиксировал закономерность развития симптомов
- Синдром Эдвардса затрагивает больше женский пол, чем мужской - около 80 % пострадавших составляют женщины
- Женщины старше тридцати лет имеют больший риск рождения ребенка с синдромом
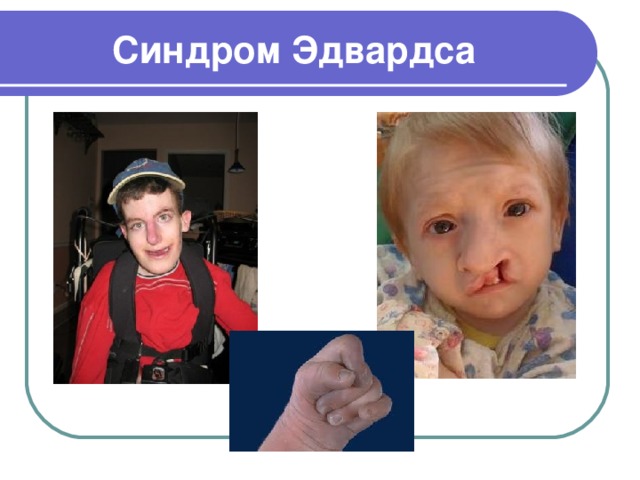
Синдром Эдвардса

Синдром Орбели
- Синдром Орбели обусловлен делецией (потеря участка хромосомы) длинного плеча тринадцатой хромосомы
- Популяционная частота синдрома не установлена
- Дети с синдромом Орбели рождаются с низким (2200 г) весом
- Клинически синдром проявляется аномалиями развития всех систем организма, характерны микроцефалия (уменьшение размеров черепа), отсутствие носовой вырезки (лоб непосредственно переходит в нос), широкая спинка носа, высокое нёбо, низко расположенные деформированные ушные раковины, поражения глаз, опорно-двигательного аппарата, часты пороки развития сердца, почек, головного мозга
- Для всех детей с синдромом Орбели характерна глубокая олигофрения, возможны потери сознания и судороги
- Большинство больных с синдромом погибают на 1-м году жизни

Синдром Орбели

Синдром Патау
- Синдром Патау - хромосомная аномалия, представляющая собой трисомию по 13-ой паре аутосом
- Синдрома Патау также встречается в литературе под названиями трисомия D и трисомия 13
- Кариотип 47, ХХ, 13+ или 47, ХУ, 13+
- Частота рождения детей с синдромом Патау составляет 1:7000-10000; соотношение полов примерно одинаковое
- Связь заболевания с увеличением количества хромосом 13-ой пары была установлена в 1960 г. К. Патау, по имени которого данный синдром и получил свое название
- При синдроме Патау у ребенка имеются множественные и крайне тяжелые аномалии развития, определяющие частые случаи внутриутробной гибели плода и малую продолжительность жизни детей с данной патологией
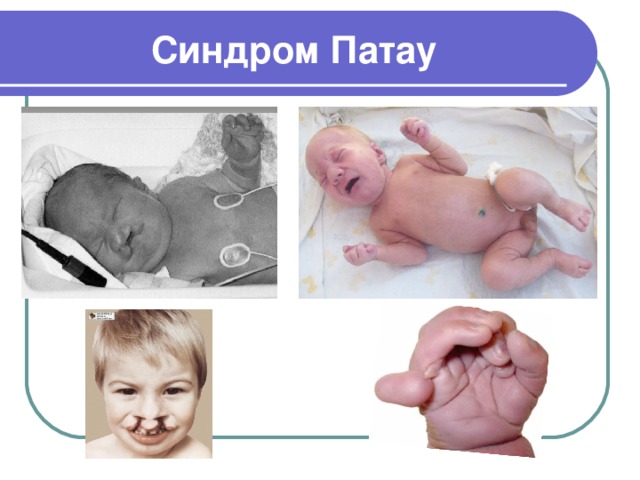
Синдром Патау

Синдром Шерешевского-Тернера
- Синдром Шерешевского-Тернера – это хромосомное заболевание, вызванное отсутствием или дефектом одной Х-хромосомы
- Кариотип 45,Х0
- В клетках отсутствуют тельца полового хроматина
- Частота 1:2000-1:5000
- Синдром описали русский клиницист М. А. Шерешевский (1925) и Г. Тернер (1938)
- Клинические диагностические признаки: женский фенотип; низкий рост, короткая шея с латеральными складками кожи (шея сфинкса), низкая граница роста волос на затылке, грудная клетка щитообразной формы с широко расставленными сосками, бесплодие
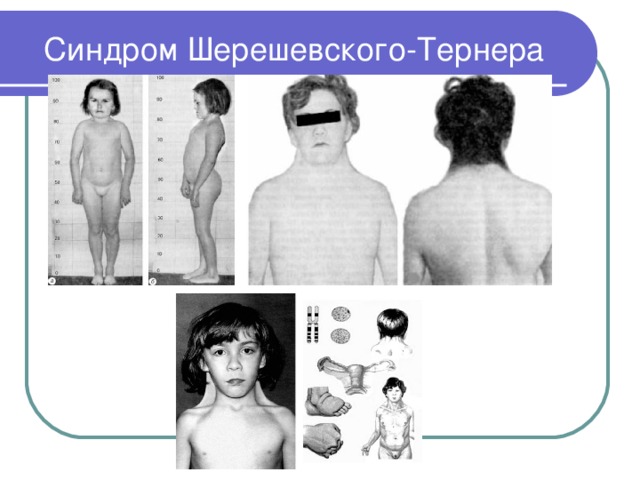
Синдром Шерешевского-Тернера

2. Генные (молекулярные) болезни
- Генные болезни – это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена
- Большинство генных патологий обусловлено мутациями в структурных генах, осуществляющих свою функцию через синтез полипептидов – белков. Любая мутация гена ведет к изменению структуры или количества белка
- В результате мутации гена на молекулярном уровне возможны следующие варианты:
- синтез аномального белка
- выработка избыточного количества генного продукта
- отсутствие выработки первичного продукта
- выработка уменьшенного количества нормального первичного продукта

Генные (молекулярные) болезни:
- Гемофилия А
- Гемофилия В
- Полидактилия
- Альбинизм
- Фенилкетонурия
- Катаракта врожденная
- Синдром Марфана
- Синдром Коффина-Лоури
- Нейрофиброматоз
- Синдром Морриса
- Талассемия
- Галактоземия
- Прогерия

Гемофилия А
- Гемофилия А (классическая гемофилия) – генетическое заболевание, вызванное врожденным дефицитом белка фактора свертывания крови VIII
- Наиболее часто встречающаяся форма гемофилии (около 80 % случаев)
- Заболевание связано с рецессивной мутацией в X-хромосоме. Встречается у мужчин и у гомозиготных женщин
- Гемофилия встречается примерно у 1 из 5000 мужчин. Из них 85 % имеют гемофилию А и 15 % - гемофилию B
- У пациентов с гемофилией может наблюдаться различный уровень активного фактора свертываемости крови: менее чем 1 % активного фактора – тяжелая форма гемофилии, 1-5 % активного фактора относится к умеренной гемофилии, а легкая форма гемофилии характеризуется диапазоном 5-40 % от нормального уровня активного фактора свертывания крови
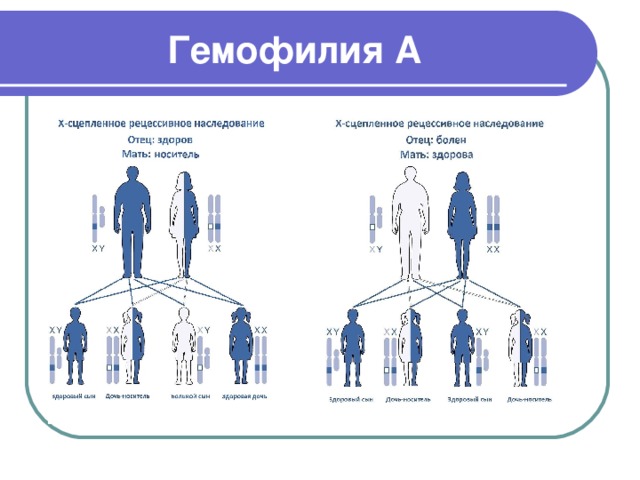
Гемофилия А
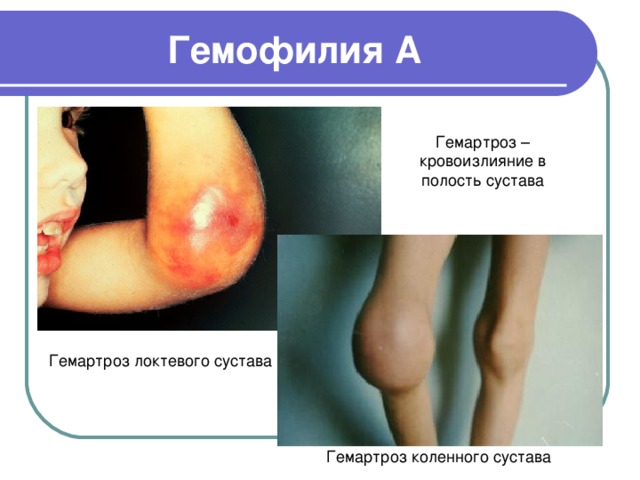
Гемофилия А
Гемартроз – кровоизлияние в полость сустава
Гемартроз локтевого сустава
Гемартроз коленного сустава

Гемофилия В
- Гемофилия B (болезнь Кристмаса) – наследственное заболевание крови, обусловленное недостаточностью коагуляционного фактора IX, основными проявлениями которого являются рецидивирующие кровотечения
- Частота появления гемофилии B составляет один на 25000-30000 новорожденных мальчиков. При этом гемофилия B встречается примерно в 5 раз реже гемофилиии А
- Наследование при этом заболевании сцеплено с полом
- Основные клинические: кровотечения различных локализаций, рецидивирующие геморрагии (кровоизлияния) в суставы и мышцы, кровотечения после любых, даже небольших травм и хирургических вмешательств
- Наиболее проблемным проявлением этого заболевания является гемофилическая артропатия, которая обусловлена рецидивами гемартрозов

Гемофилия В
Основным и наиболее эффективным способом лечения кровотечений у пациентов, страдающих гемофилией В, является пожизненная заместительная терапия концентратами коагуляционного фактора IX

Полидактилия
- Полидактилия – одна из врожденных аномалий развития, проявляющаяся увеличением количества пальцев на руках или ногах. Полидактилию еще называют многопалостью
- Встречается она с одинаковой частотой у девочек и у мальчиков, 1 случай на 600-3500 новорожденных
- Главная причина этой патологии – наследственный дефект. Учеными установлено, что полидактилия передается по наследству
- Тип наследования – аутосомно-доминантный, но пенетрантность (вероятность появления признака) составляет 50 %, то есть у здоровых родителей может родиться ребенок с полидактилией
- Главный признак полидактилии – наличие дополнительных пальцев на кистях или стопах. Эти пальцы могут иметь рудиментарный вид, когда они состоят только из мягкой ткани и кожи, а могут быть и полноценными пальцами. Чаще всего добавочные пальцы деформированы, имеют меньшее число фаланг
- При симптоматической полидактилии у ребенка часто отмечаются и другие аномалии развития – врожденные пороки сердца, укорочение конечностей, деформация ушных раковин и прочее
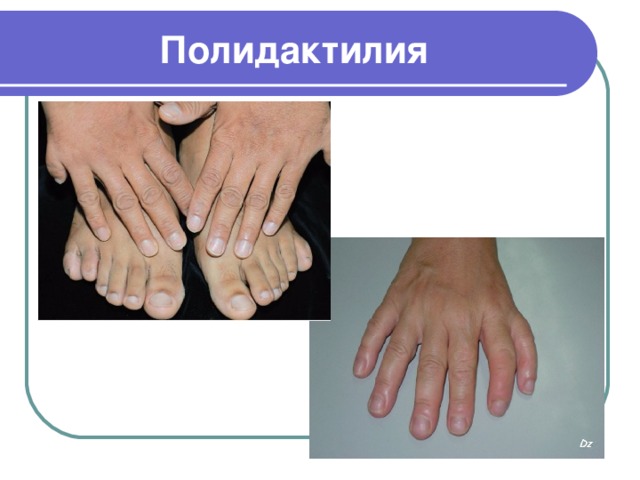
Полидактилия

Альбинизм
- Альбинизм является врожденным заболеванием. Эта болезнь подразумевает отсутствие в коже, волосах, ногтях, пигментной и радужной глазных оболочках пигмента
- Возникает альбинизм из-за отсутствия или блокады фермента тирозиназы. Он крайне важен для выработки меланина
- Альбинизм наследуется от родителей. Он появляется у ребенка в том случае, если оба родителя являются носителями дефектного гена.
- Когда дефектный ген присутствует только у одного родителя, альбинизм у детей не развивается, но в организме все равно остается мутировавший ген, который может передаться следующему поколению (аутосомно-рецессивное наследование)

Альбинизм

Фенилкетонурия
- Фенилкетонурия - это наследственное заболевание, которое характеризуется нарушением белкового обмена
- Впервые это заболевание обнаружили в 1934 году
- Фенилкетонурия наследуется по аутосомно-рецессивному типу, то есть у совершенно здоровых родителей (носителей) могут родиться больные фенилкетонурией дети
- Основными причинами развития фенилкетонурии у детей считаются мутации гена, находящегося на 12 хромосоме
- Существует 3 типа этого заболевания:
- фенилкетонурия первого типа –дефицит в организме фермента фенилаланин-4-гидроксилаз . Чаще всего наследуется фенилкетонурия именно этого типа (в 98% случаев);
- фенилкетонурия второго типа – недостаток фермента дигидроптеридинредуктаза . Больные страдают судорогами и умственной отсталостью. Этот тип фенилкетонурии встречается довольно редко (1-2%), но обычно приводит к смертельному исходу в 2-3-летнем возрасте;
- фенилкетонурия третьего типа – характеризуется дефицитом тетрагидробиоптерин а. Симптомы: умственная отсталость вследствие микроцефалии – уменьшения объема мозга
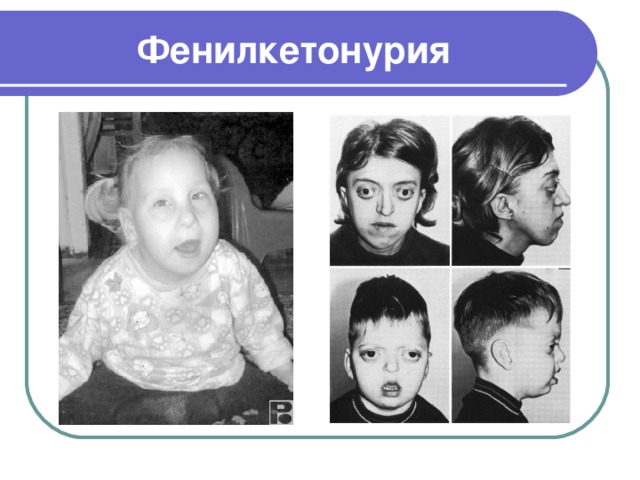
Фенилкетонурия

Катаракта врожденная
- Катаракта врожденная – заболевание, при котором природный хрусталик потерял свою прозрачность и имеет помутнения
- Врожденная катаракта развивается по многим причинам: наследственные факторы, инфекционные и воспалительные заболевания, нарушения обмена веществ, сахарный диабет, использование некоторых лекарственных препаратов
- Например, использование антибиотиков тетрациклинового ряда при лечении инфекций у беременных женщин может стать причиной развития катаракты у новорожденных младенцев
- Генетические изменения в структуре белков, необходимых для обеспечения прозрачности хрусталика, обуславливают развитие врожденной катаракты
- Врожденные катаракты составляют до 60 % всех аномалий развития глазного яблока и встречаются примерно у 0,4 % новорожденных детей
- Большинство врожденных катаракт являются показанием к операции, но в некоторых случаях, когда помутнение в хрусталике находится на периферии и не снижает центральное зрение, хирургическое лечение не требуется
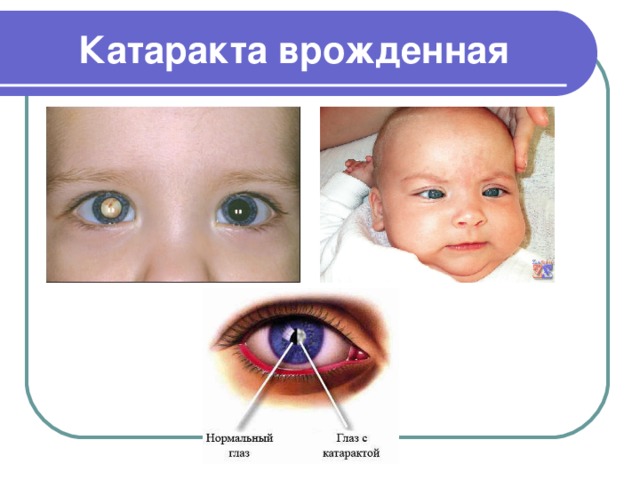
Катаракта врожденная

Синдром Марфана
- Синдром Марфана (или арахнодактилия) – это наследственное заболевание, характеризующееся недостаточностью соединительной ткани
- Синдром Марфана обусловлен мутацией в 15-й хромосоме гена белка фибриллина, вследствие чего нарушается структура и выработка коллагена
- Болезнь приводит к патологическим изменениям сердечно-сосудистой, нервной, опорно-двигательной и других систем и органов
- Арахнодактилия наследуется по аутосомно-доминантному типу, поэтому встречается почти в одинаковом соотношении, как у мужчин, так и у женщин
- Это достаточно редкое генетическое заболевание с частотой возникновения 1:5000
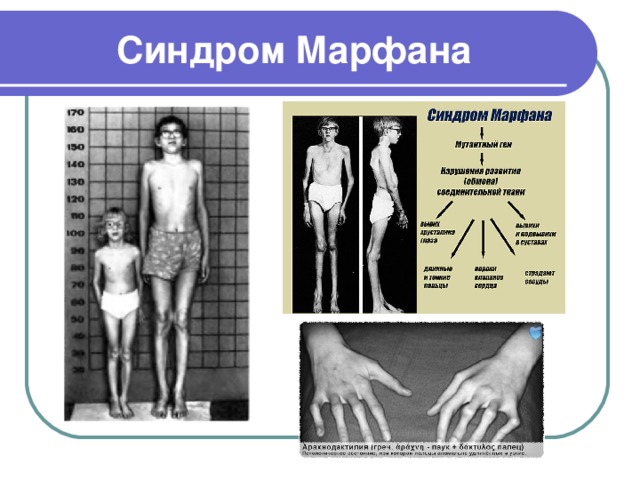
Синдром Марфана

Синдром Коффина-Лоури
- Синдром Коффина-Лоури – редкая форма Х-сцепленной умственной отсталости, сопровождающаяся изменениями скелета, задержкой роста, нарушением слуха
- Причина: мутации в гене, кодирующем белок, участвующий в регуляции клеточного цикла
- Соотношение полов – M1:Ж1
- Тип наследования – Х-сцепленное доминантное
- Для больных характерны грубые черты лица, широкий выступающий лоб, гипертелоризм (увеличенное расстояние между внутренними краями глазниц), густые брови, большой открытый рот с толстой оттопыренной нижней губой, большие вытянутые вверх уши

Синдром Коффина-Лоури

Нейрофиброматоз
- Нейрофиброматоз (или болезнь Реклингхаузена) – заболевание генетически-наследственное, которое влияет на все клетки нервного гребня и относится к определенной группе патологий – факоматозов
- Такая патология обусловлена нарушением обмена в организме в целом, которое вызвано повреждением гена, отвечающего за синтез какого-либо фермента
- Тип наследования аутосомно-доминантный
- Частота возникновения заболевания у мужчин и женщин одинакова, наблюдается примерно у каждого 3500 новорожденного
- Данное заболевание поражает многие органы и даже целые системы. Возможны пороки развития кожи, нервной системы, глаз, внутренних органов и т. д.
- Внешне он представляет собой множество пятен на коже кофейно-молочного цвета, нейрофибром и пигментных гамартом радужки (узелки Леща)
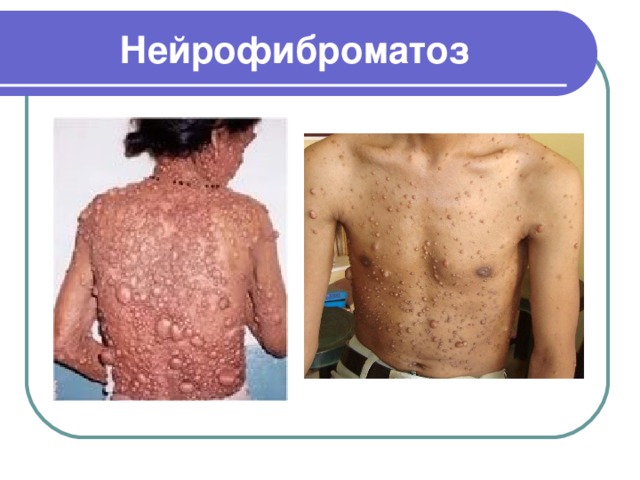
Нейрофиброматоз

Синдром Морриса
- Синдром нечувствительности к андрогенам (синдром тестикулярной феминизации) – врожденные эндокринные нарушения полового развития, вызванные мутацией гена, отвечающего за андрогеновый рецептор
- Синдром наследуется с Х-хромосомой как рецессивный признак
- Проявляется нарушениями полового развития, которые развиваются в результате слабого реагирования на мужские половые гормоны у лиц с мужским набором хромосом (XY)
- Данный синдром является наиболее известной причиной развития мужчины как девушки или наличия проявлений феминизации у мальчиков, которые родились с мужским набором хромосом и нормальным уровнем половых гормонов
- Частота заболеваемости, примерно 1-5 на 100000 новорожденных

Синдром Морриса

Талассемия
- Талассемия (анемия Кули) – заболевание, наследуемое по рецессивному типу, в основе которого лежит снижение синтеза полипептидных цепей, входящих в структуру нормального гемоглобина
- Талассемию вызывают точечные мутации или делеции (потеря участка хромосомы) в генах гемоглобина, ведущие к нарушению синтеза РНК, что приводит к уменьшению или полному прекращению синтеза одного из видов полипептидных цепей. Синтез цепей другого вида продолжается. Это приводит к образованию нестабильных полипептидных агрегатов из избыточных цепей, нарушающих нормальное функционирование эритроцитов и их разрушению
- При талассемии характерны гипохромная анемия анизоцитоз эритроцитов (изменение размеров), нарушения в строении лицевого черепа (квадратный, башенный); нос приобретает седловидную форму; нарушается прикус и расположение зубов. Отмечается желтушность кожи и слизистых оболочек. Селезенка и печень увеличены. Больные подвержены инфекционным заболеваниям. Рано начавшаяся анемия обуславливает физическое и умственное недоразвитие ребёнка
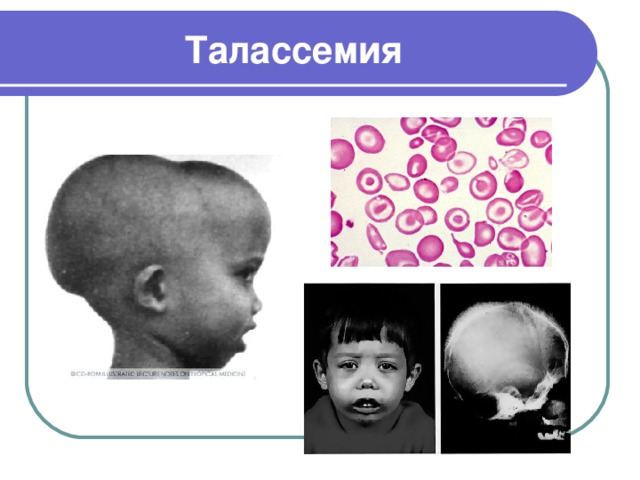
Талассемия

Галактоземия
- Галактоземия – редкое наследственное заболевание, вызванное нарушением обмена веществ, при котором происходит ненормальный процесс метаболизма углеводов галактозы
- Возникает в результате врожденного дефекта определенных генов Галактоземия у новорожденных встречается примерно в 1 случае из 15-20 тысяч
- Первые симптомы галактоземии у ребенка возникают уже спустя пару суток после рождения: возникают они на фоне кормления молочной пищей и проявляются как рвота и расстройства стула, кишечная колика и вздутие живота, появляется желтуха и обильное выделение газов
- При отсутствии своевременной диагностики галактоземии у новорожденных увеличивается печень в размере и развивается поражение нервной системы – снижение мышечного тонуса, судороги
- Постепенно симптомы галактоземии проявляются в выраженном отставании в психическом и физическом развитии, может наблюдаться помутнение хрусталика (катаракта). Главной же проблемой является цирроз печени, который при отсутствии адекватного лечения является основной причиной летального исхода
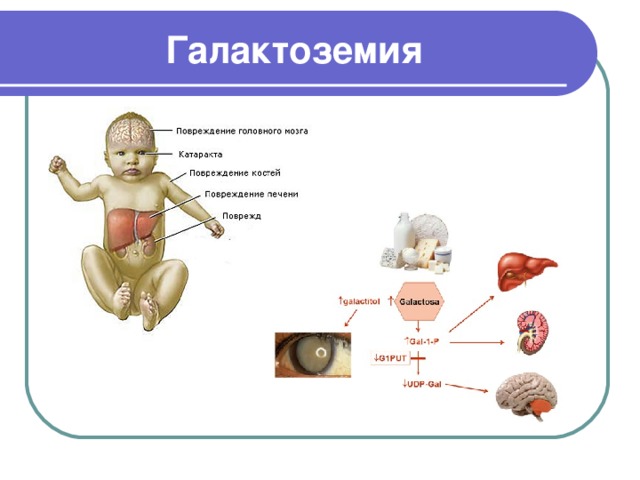
Галактоземия

Прогерия
- Прогерия – редкое генетическое заболевание, впервые описанное Гилфордом, которое проявляется в преждевременном старении организма, связанное с его недоразвитием
- Причина детской прогерии – мутации гена LMNA, кодирующего ламин А.
- Ламины – белки, из которых выстроен особый слой оболочки клеточного ядра
- Прогерия взрослых имеет аутосомно-рецессивный тип наследования
- При прогерии возникают изменения кожи и внутренних органов
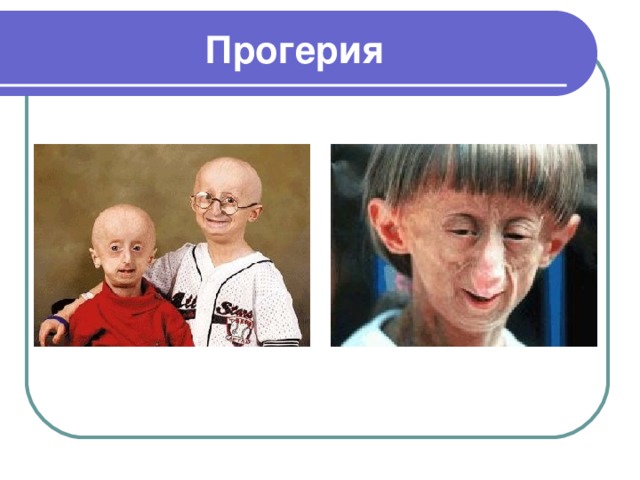
Прогерия

3. Мультифакториальные заболевания
- Мультифакториальные заболевания (наследственно предрасположенные, многофакторные – это заболевания, развитие которых определяется взаимодействием определенных наследственных факторов (мутаций или сочетаний аллелей) и факторов среды
- Мультифакториальные болезни можно разделить на:
1) врожденные пороки развития,
2) распространенные психические и нервные болезни,
3) распространенные болезни «среднего» возраста
- Примеры заболеваний: бронхиальная астма, язвенная болезнь, сахарный диабет, ишемическая болезнь сердца, псориаз, шизофрения

Спасибо за внимание!








Вебинар для учителей

Свидетельство об участии БЕСПЛАТНО!

